III ) 3 Influence de l’additif A (Base) sur la réduction des espèces manganèse
III ) 3.1 Etude en voltamétrie cyclique sur carbone vitreux et sur acier
III ) 3.1.1 En absence d'additif
La figure 18 présente la partie cathodique des voltamogrammes obtenue sur carbone vitreux et sur acier dans une solution de chlorure de manganèse à la vitesse de 20 mVs-1. Sur carbone vitreux (courbe 18a), aucun courant n'est enregistré jusqu'au potentiel E=-1580 mV/ecs où se produit une augmentation rapide du courant. La présence d’un pic anodique lors du balayage retour indique la formation d’un dépôt de manganèse. La présence de ce pic est d'autant plus intéressante qu’elle permet de déterminer le mode de croissance du dépôt. En effet, nous observons, d'une part, que la valeur du courant enregistrée lors du balayage retour est supérieure à celle du courant aller. D'autre part, nous savons que le carbone vitreux ne permet pas la formation d'un dépôt par des mécanismes autres que la germination et croissance en trois dimensions. Ainsi sur carbone vitreux, la cristallisation du manganèse se produit par un mécanisme de germination tridimensionnelle. De plus, cette germination intervient avec une surtension de 155 mV par rapport au potentiel d’équilibre du système manganèse qui est déterminé sur la courbe par le passage au potentiel à courant nul E(i=0)=-1425 mV/ecs. Le changement de pente intervenant aux potentiels cathodiques par rapport à –1580 mV/ecs est la conséquence du démarrage de l’évolution de l’hydrogène sur des cristallites de manganèse nouvellement formées.
Sur acier, le voltamogramme 18b obtenu est complètement différent. Contrairement au cas précédent, le courant prend, dès le début de la polarisation, une valeur non nulle. Il se forme un large pic MH dans une zone de potentiel anodique par rapport au potentiel d’équilibre du manganèse.
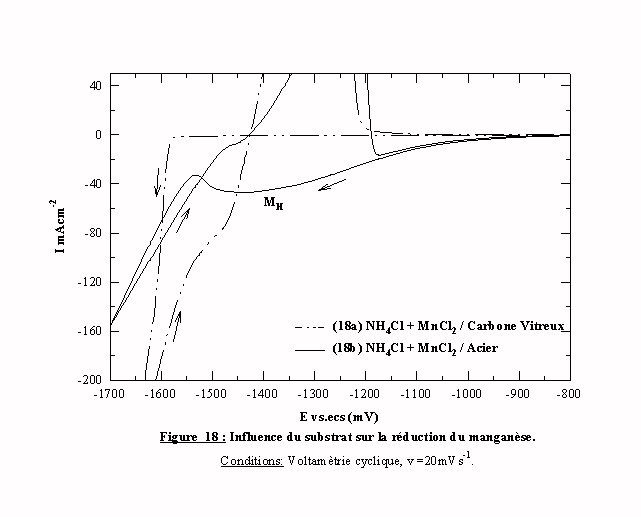
Ce large pic est néanmoins suivi d’une augmentation de courant pour des potentiels cathodiques par rapport à E=-1530 mV/ecs. Le passage à courant nul du balayage retour se fait au même potentiel que sur le substrat de carbone vitreux, c'est à dire au potentiel d'équilibre du manganèse.
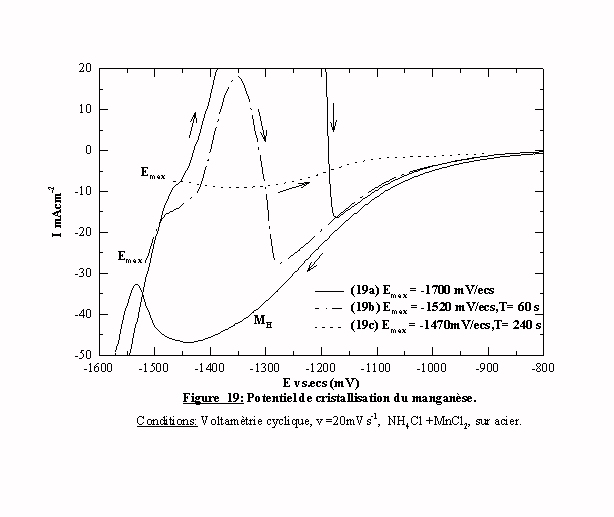
Afin de déterminer précisément le potentiel de réduction des espèces manganèse sur l’acier, nous avons inversé le sens du balayage à différents potentiels (Figure 19). Cependant il est connu que le manganèse se dissout chimiquement dans le chlorure d'ammonium. En effet, dans une solution acide, le manganèse est chimiquement corrodé par un mécanisme à deux étapes dont la première étape est la formation d'un oxyde à la surface [50 et ses références]:
![]()
Mn + H2O è MnO + H2
MnO +2H+ è Mn2+
+ H2O
Les résultats expérimentaux de G. Provoost [50] montrent qu'une autre réaction se produit simultanément:
MnO + NH4+ è MnOH+
+ NH3
(Remarque: Cette réaction parallèle est contrôlée par transfert de masse de NH4+ à la surface de l'électrode.)
Cette dissolution rapide peut entraîner la disparition du pic anodique de dissolution si la quantité de manganèse déposée est trop faible. Pour s’affranchir de ce risque, nous choisissons d'imposer un temps d'attente suffisamment long au potentiel maximum afin de déposer une quantité de manganèse suffisante pour observer un pic anodique. Quand le délai au potentiel maximum E=-1520 mV/ecs est de 60 secondes, la courbe (19b) présente un pic anodique attestant de la cristallisation du manganèse. Ce balayage retour présente également d’autres caractéristiques. On note successivement la présence d’une inflexion, du pic anodique puis d’une réactivation cathodique. L'inflexion est la conséquence probable de l'arrêt de l'évolution de l'hydrogène. La réactivation correspond au redémarrage de la réduction de l’hydrogène sur l’acier. En effet on retrouve le tracé de la courbe (19a). Par contre avec un délai au potentiel maximum E=-1470 mV/ecs de 240 secondes, la courbe (19c) montre un balayage retour sans pic anodique dont l'intensité du courant reste très inférieure à celle du balayage aller. Le potentiel de réduction des espèces manganèse est donc cathodique par rapport à cette valeur. Le potentiel de réduction des espèces manganèse est donc compris entre –1470 et –1520 mV/ecs.
Dépôt en sous potentiel de
manganèse
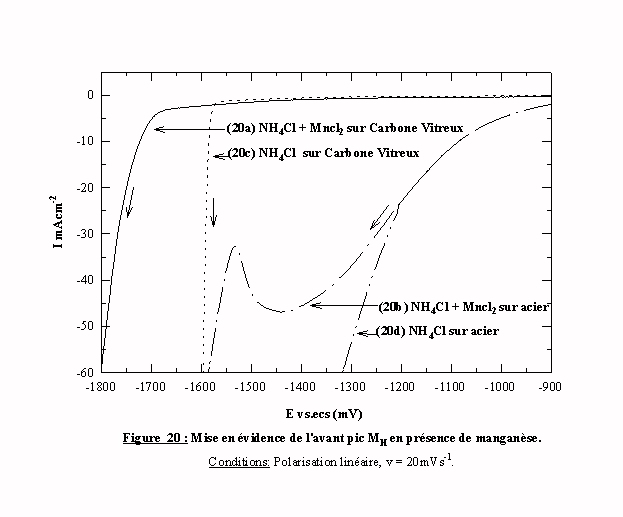
Pour examiner plus précisément le pic MH , les balayages aller de la courbe 18 sont représentés sur la même figure. Les courbes d’évolution de l’hydrogène sur l’acier (courbe (20d)) et sur le carbone vitreux (courbe (20c)) sont également reportées. La courbe (20c) montre la très grande surtension de l’évolution de l’hydrogène sur le carbone vitreux qui intervient à un potentiel plus cathodique que la réduction des espèces manganèse ce qui confirme l’hypothèse proposée pour la cassure de pente enregistrée sur la courbe (18a). La similitude des courbes (20b) et (20d) entre -900 mV/ecs et -1200 mV/ecs montre que le large pic MH correspond à l’évolution de l’hydrogène sur l’acier nu. Cette évolution subit ensuite un blocage comparable à celui déjà observé pour le zinc lors du premier pic ZH. Bien que cette zone de potentiel soit plus anodique par rapport au potentiel d'équilibre du système Mn/Mn2+, ce blocage est imputable à l'introduction des espèces manganèse comme dans le cas du zinc (cf. § III) 2.1 ). Deux réactions de réduction se superposent dans la même zone de potentiel: un dépôt en sous potentiel de manganèse et la réduction du proton. Comme dans le cas du zinc, c'est l'UPD de manganèse qui ralentit puis bloque partiellement la réduction de l'hydrogène. L'occupation des sites électroactifs par les espèces Mnads est également mise en évidence par une expérience de perméation.
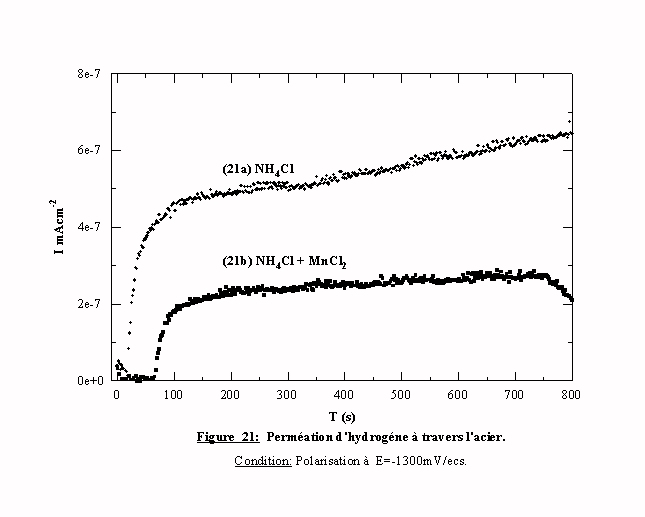
L'étude de la perméation de l'hydrogène, dans la région d'UPD est effectuée avec une polarisation à -1500 mV/ecs de la cellule de chargement contenant du chlorure d'ammonium (transitoire 21a) et du chlorure d'ammonium avec des espèces manganèse (transitoire 21b). Dans les deux cas, les tracés présentent la même allure générale. Dès que la polarisation s'établit, le courant augmente rapidement jusqu'à l'établissement d'un courant stationnaire de perméation. Le chlorure d'ammonium donne une valeur de courant stationnaire proche de 0,50 mAcm-2, en présence de manganèse cette valeur est proche de 0,25 mAcm-2. Le manganèse agit donc directement à la surface puisqu'il modifie le taux de recouvrement de l'hydrogène adsorbé, il y a bien une compétition entre les espèces Hads et Mnads pour l'occupation des sites électroactifs. Cet essai confirme l'hypothèse d'un dépôt en UPD de manganèse.
III ) 3.1.2 En présence d'additif
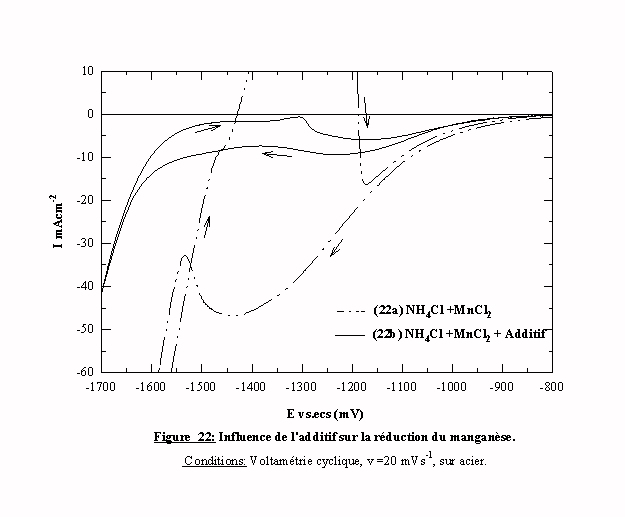
En présence d’additif, l’allure du voltamogramme change, mais conserve des caractéristiques générales qui permettent de la comparer à celle du voltamogramme obtenue sans additif (figure 22). Les courants qui apparaissent en présence d'additif (courbe 22b) restent cependant nettement inférieurs à ceux de la courbe (22a). Dès le début de la polarisation, la réduction de l'hydrogène est fortement atténuée. De plus, le pic de réduction des espèces manganèse débute avec une surtension de 100 mV par rapport à la solution sans additif. Ainsi les caractéristiques induites par l’introduction d'additif sont comparables à celles obtenues dans le cas de l'électrolyte contenant du zinc (cf. § III) 2 .1 ). L’additif joue le même rôle de blocage des sites actifs qui neutralise partiellement la réduction de l’hydrogène, l’UPD et la réduction des espèces manganèse. L’aspect le plus important du voltamogramme 22b est la disparition du pic anodique lors du balayage retour. En effet, elle peut signifier que l'additif bloque totalement la réduction des espèces manganèse. Cette absence de dépôt de manganèse en présence d’additif semble hypothéquer la possibilité d’obtenir un codépôt de zinc-manganèse, à moins d'obtenir une codéposition anormale (cf. § I) 4.4 ) induite par la présence des espèces zinc. Nous pouvons grouper les résultats de l'étude en voltamétrie cyclique dans le tableau suivant:
|
Potentiel en mV/ecs |
MnCl2 + NH4Cl /acier |
MnCl2 + NH4Cl + Additif
/acier |
|
-750 < E < -1200 |
Réduction de H+ |
Blocage partiel de la réduction de H+ |
|
-1200 < E <-1520 |
UPD de Manganèse |
Blocage de l'UPD de Manganèse |
|
-1520 < E < -1620 |
Réduction de Mn2+ et H+ |
Blocage de l'UPD de Manganèse |
|
-1620< E <-1700 |
Réduction de Mn2+ et H+ |
Réduction de Mn2+et H+ sous influence de l'additif |
|
Contrôle cinétique |
Transfert de charge |
Adsorption de l'additif |
|
Mode de croissance |
Germination 3D sur C.V Stranski Krastanov sur acier* |
Stranski Krastanov sur acier* |
|
Pic anodique |
Caractéristique dissolution rapide |
Absent |
* Cf. Polarisation potentiostatique
III ) 3.2 Etude en polarisation potentiostatique
III ) 3.2.1 En absence d'additif
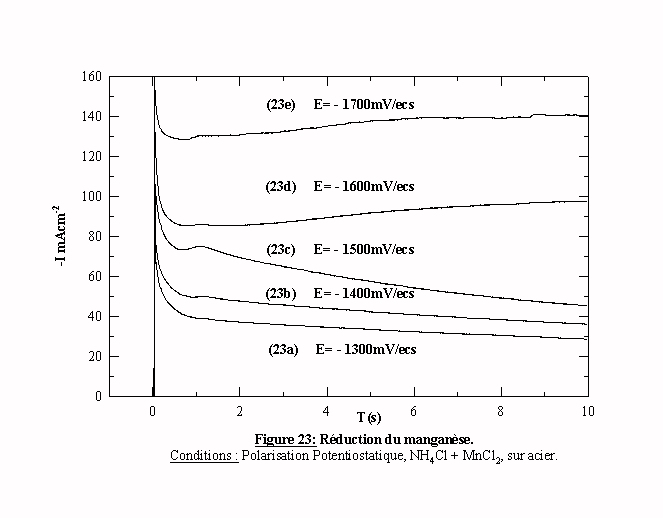
La figure 23 présente les transitoires obtenus dans une solution contenant le chlorure d'ammonium et le chlorure de manganèse. En fonction des potentiels appliqués, nous observons trois types de transitoires.
a) Quand le potentiel appliqué est anodique par rapport au potentiel où se produit l'UPD, le transitoire 23a (E=-1300 mV/ecs) présente une décroissance monotone caractéristique de l'évolution de l'hydrogène sur l'acier.
b) Quand le potentiel appliqué est cathodique par rapport à cette valeur, les transitoires 23b (E=-1400 mV/ecs) et 23c (E=-1500 mV/ecs) présentent une décroissance puis un maximum suivi par une nouvelle décroissance de courant. La valeur du courant maximum Im augmente quand le potentiel devient de plus en plus cathodique, sans variation du temps tm mis pour atteindre cette valeur.
c) Quand la valeur du potentiel imposé est cathodique par rapport à -1520 mV/ecs, les transitoires 23d et 23e montrent une augmentation de l'intensité du courant après le petit pic. Cette augmentation correspond au processus de nucléation du manganèse.
Lorentz [25] a étudié les mécanismes d'électrocristallisation en présence d'une couche adsorbée. Il obtient, en simulant la simultanéité de trois processus différents, le même type de transitoire. Ces trois processus concernent :
![]() La formation d'une
monocouche bidimensionnelle (2D) ordonnée des espèces adsorbées, à un potentiel
anodique par rapport au potentiel d'équilibre (UPD).
La formation d'une
monocouche bidimensionnelle (2D) ordonnée des espèces adsorbées, à un potentiel
anodique par rapport au potentiel d'équilibre (UPD).
![]() La transition de la
phase 2D vers une phase bidimensionnelle (3D) qui forme les germes de
cristallisation du dépôt de manganèse.
La transition de la
phase 2D vers une phase bidimensionnelle (3D) qui forme les germes de
cristallisation du dépôt de manganèse.
![]() La croissance du dépôt
pour un potentiel cathodique par rapport au potentiel d'équilibre.
La croissance du dépôt
pour un potentiel cathodique par rapport au potentiel d'équilibre.
En transposant les résultats de cette étude, nous pouvons dire que le manganèse cristallise en suivant un mode de croissance du type Stranski-Krastanov (cf. § I) 4.3 ). La formation de cristallites 3D intervient sur un substrat modifié par une couche adsorbée 2D.
III ) 3.2.2 En présence d'additif
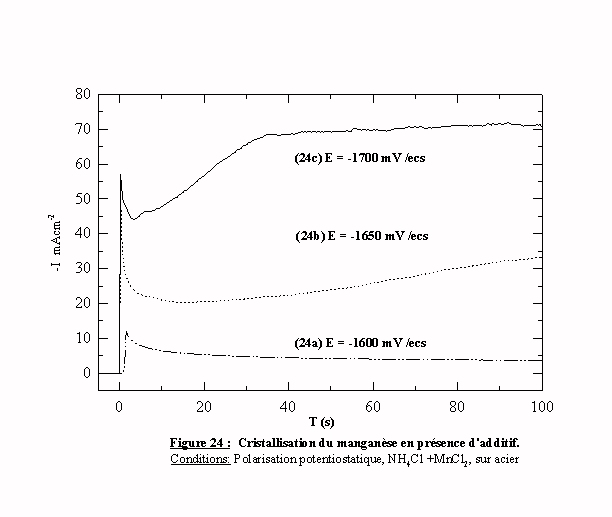
Les transitoires
obtenus en présence d'additif sont présentés sur la figure 24. Pour un potentiel
E=-1600 mV/ecs, le transitoire 24a présente une décroissance monotone qui
correspond à l'évolution de l'hydrogène puis une stabilisation à une densité de
courant inférieure à 5 mAcm-2. Donc, pour des potentiels anodiques
par rapport à –1600 mV/ecs, l'additif bloque totalement la cristallisation du
manganèse. Pour des potentiels plus cathodiques, les transitoires 24b et 24c
présentent une décroissance, puis une augmentation de courant avec des
intensités de courant enregistrées très inférieures à celles enregistrées sans
additif. Sur le transitoire 24c, le petit pic après la décroissance est encore
observé. L'allure caractéristique du tracé semble indiquer que le manganèse
commence à cristalliser en suivant le même mode qu'en absence d'additif. Cependant
nous n'avons jamais pu obtenir de dépôt de manganèse sur l'acier en présence
d'additif. Cet aspect peut signifier que l'additif bloque la croissance du
dépôt de manganèse en s'adsorbant plus efficacement sur le manganèse que sur
l'acier. La vérification de cette hypothèse constitue à elle seule un sujet
d'étude que nous ne pouvons pas aborder. Nous pouvons simplement dire que pour
des potentiels cathodiques par rapport à –1600 mV/ecs, l'additif induit une
surtension ou bloque la cristallisation du manganèse.
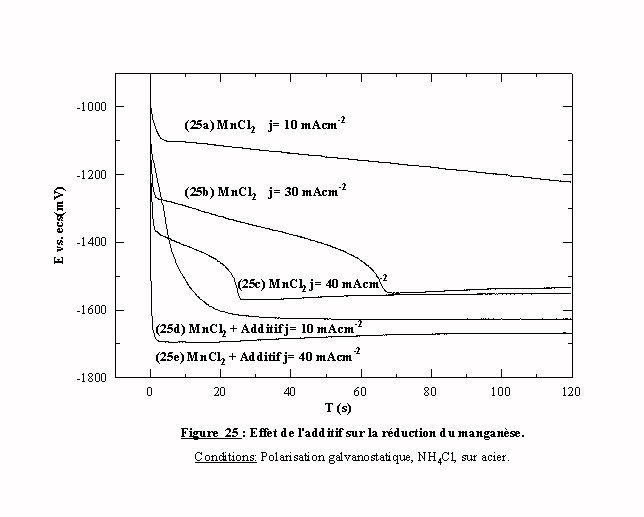
III ) 3.3 Etude en polarisation galvanostatique
La figure 25 présente les transitoires potentiel-temps d'un bain de chlorure de manganèse avec et sans additif. Sans additif, les transitoires 25a, 25b , 25c sont obtenus pour des courants respectivement de 10, 30 et 40 mAcm-2. Ces transitoires présentent une décroissance du potentiel en trois étapes au cours du temps, jusqu’à une stabilisation proche de -1550 mV/ecs qui correspond au potentiel de réduction des espèces manganèse. Chaque système atteint ce potentiel d’équilibre avec un délai qui est directement fonction du courant imposé. Le temps mis pour atteindre un potentiel est plus court pour les forts courants.
En présence d’additif, l’allure des transitoires est différente: la décroissance est rapide, régulière et monotone, la valeur du potentiel atteint très rapidement des valeurs inférieures à –1600 mV/ecs. L’additif impose donc très rapidement un potentiel cathodique par rapport au potentiel de réduction des espèces manganèse seul. Cependant nous ne pouvons pas dire si la cristallisation du manganèse se produit en présence d’additif.
III ) 3.4 Conclusion
Sur acier, le manganèse cristallise pour des potentiels cathodiques par rapport à –1500 mV/ecs en suivant le mode de croissance Stranski-Krastanov. La cristallisation est précédée d'une adsorption en sous potentiel. Le système est placé sous contrôle de transfert de charge. L'introduction de l'additif induit une surtension à la cristallisation du manganèse.
III ) 4 Etude du bain zinc-manganèse
Nous connaissons maintenant les principaux paramètres cinétiques des deux bains contenant les espèces zinc et manganèse. Ces résultats constituent un support pour l'étude du bain de zinc-manganèse en présence et en absence d'additif. La première partie de cette étude suit la même méthodologie en utilisant la voltamétrie cyclique, la polarisation galvanostatique et potentiostatique, sur substrat d'acier.
III ) 4.1 Etude en voltamétrie cyclique
III ) 4.1.1 En absence d'additif
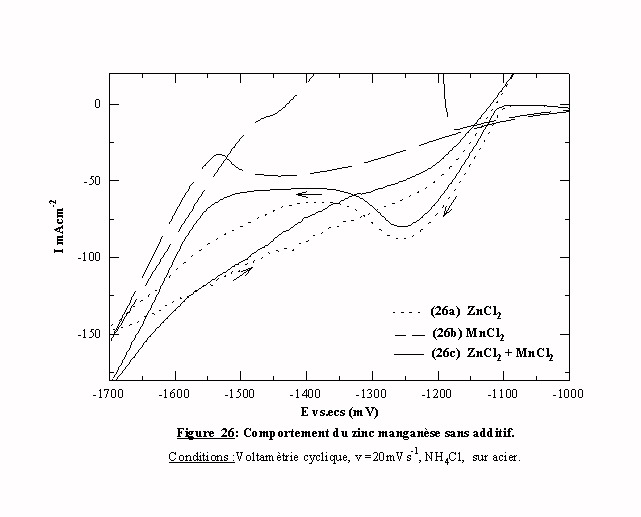
La figure 26 présente le voltamogramme (26c) d'une solution de chlorure de zinc et de manganèse à la vitesse de 20 mVs-1 sur l'acier. Pour mémoire, les voltamogrammes des solutions ZnCl2 + NH4Cl (26a) et MnCl2+ NH4Cl (26b) sont reportés sur cette figure. Le voltamogramme 26c présente une grande similitude avec le voltamogramme (26a) ne contenant que le zinc. On remarque simplement un décalage entre les deux courbes qui se résume à une surtension de 10 mV ou à une diminution de courant de 10 mAcm-2. Le premier pic cathodique débute à un potentiel proche du potentiel de réduction du zinc (E Zn/Zn2+=-1080 mV/ecs), ce qui signifie que la réduction des espèces zinc intervient pratiquement au même potentiel qu’en l'absence de manganèse. Dans ce cas, le manganèse agit comme un élément perturbateur du système car il induit une légère surtension au système du zinc. En s'adsorbant, il neutralise une partie de la surface électroactive. En effet, cette gamme de potentiel est compatible avec un UPD de manganèse. Le second pic, qui correspond à priori à la réduction des espèces manganèse et de l'hydrogène, prend une direction parallèle à celle du voltamogramme 26b. Cependant, il reste difficile de déterminer si du manganèse se dépose. Néanmoins, s'il se produit une codéposition, celle-ci est normale et les deux éléments se déposent séparément l'un après l'autre au même potentiel que lorsqu'ils sont seuls.
III ) 4.1.2 En présence d'additif
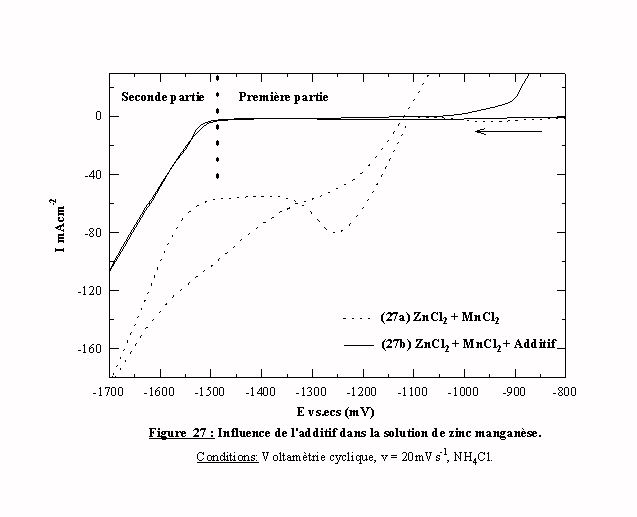
La figure 27 présente deux voltamogrammes 27b et 27a d'un bain contenant du chlorure de zinc et du chlorure manganèse respectivement avec et sans additif. En présence d’additif, le voltamogramme 27b peut se diviser en deux parties distinctes.
Pour les potentiels inférieurs à -1550 mV/ecs, l'intensité du courant mesurée est quasiment nulle. La réduction des espèces zinc et du proton est totalement bloquée par l’additif et le manganèse. Donc durant cette phase, il se dépose très peu de zinc.
Pour des potentiels supérieurs à –1550 mV/ecs, l'intensité du courant augmente. Dans cette zone de potentiels, toutes les espèces contenues dans le bain sont susceptibles d'être réduites y compris le manganèse. L’augmentation du courant se produit sans cassure de pente. Il est difficile d'envisager l'intervention de telle ou telle réaction, d'autant plus que le balayage retour suit exactement le balayage aller jusqu'au départ du pic anodique. Néanmoins le déplacement du pic vers des potentiels suffisamment cathodiques devrait favoriser la présence de manganèse dans le dépôt.
Le pic anodique comporte un changement de pente très net. L'augmentation du courant est nettement plus rapide dans la seconde partie que dans la première partie du pic. Cette caractéristique peut indiquer l'existence de deux phases issues de la combinaison du zinc et du manganèse subissant ou ne subissant pas l’influence de l’additif.
III ) 4.1.3 Influence de l'agitation en voltamétrie cyclique
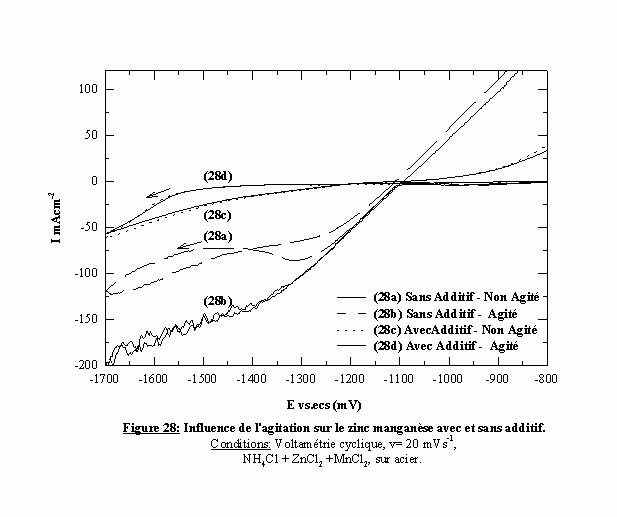
Afin de mettre en évidence le type de contrôle cinétique auquel est soumis le système zinc-manganèse, l'influence de l'agitation (figure 28) est étudiée.
Sans additif, la modification des conditions hydrodynamiques affecte la valeur du palier diffusionnel (voltamogrammes 28a 28b). Le système est placé sous contrôle cinétique de transfert de masse comme le zinc.
Par contre, en présence de l'additif (voltamogramme 28c), l'agitation n'a aucune influence (voltamogramme 28d). C'est bien l'additif qui impose la cinétique de la réaction quelques soient les conditions hydrodynamiques.
III ) 4.2 Etude en polarisation potentiostatique
La figure 29 présente des transitoires intensité-temps obtenus en présence et en absence d’additif. Les transitoires 29b et 29d présentent les résultats d’une polarisation d'une électrode d'acier à E=-1550 mV/ecs et E=-1400 mV/ecs dans un bain contenant les espèces de zinc et manganèse. Ces transitoires changent d'allure en fonction du potentiel appliqué.
Pour un potentiel anodique par rapport au potentiel de cristallisation du manganèse, le transitoire 29d garde la même allure que la solution ne contenant que l'espèce zinc (transitoire 29c), c'est à dire une décroissance puis une stabilisation à une densité de courant inférieure. On a donc la confirmation du blocage des sites électroactifs induit par l'adsorption du manganèse.
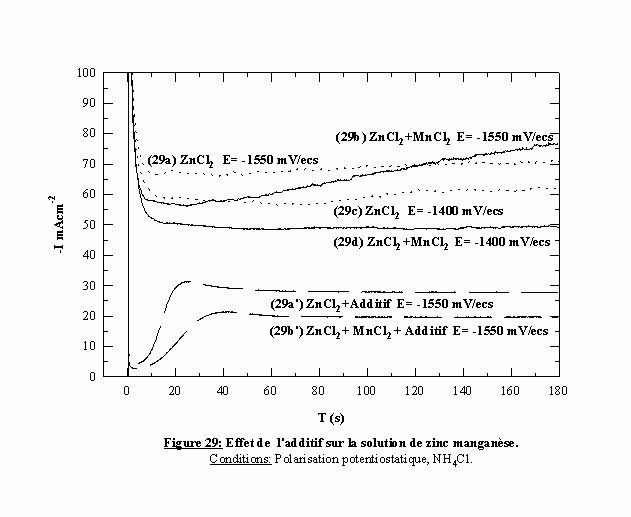
Pour un potentiel égal ou cathodique par rapport à –1550 mV/ecs, le transitoire 29b obtenu peut être divisé en deux parties. Pendant les 120 premières secondes, l'intensité du transitoire 29b est inférieure au transitoire obtenu dans le chlorure de zinc (29a). On peut penser que le manganèse bloque partiellement la réduction des espèces zinc et/ou du proton. Au cours du temps l'efficacité du blocage de ces réactions décroît jusqu'à son annulation. Après deux minutes, la valeur du courant devient supérieure à celle obtenue dans le chlorure de zinc (29a). Cette augmentation du courant peut être la conséquence, soit d'une modification du mécanisme de réduction des espèces zinc et /ou du proton, soit de la cristallisation du manganèse.
En présence d’additif, le transitoire 29b' contenant le zinc et le manganèse présente la même allure que celle du zinc en présence d’additif 29a' avec une valeur de densité de courant inférieure. Donc, même en présence d'additif le manganèse induit un blocage de la réduction des espèces zinc identique à celui obtenu sans additif.
III ) 4.3 Etude en polarisation galvanostatique
La figure 30 présente les transitoires potentiel-temps à partir des solutions contenant les espèces zinc et manganèse, avec et sans additif. Pour mémoire, les transitoires obtenus dans les solutions qui ne contiennent qu'une espèce (transitoires 30a du zinc et 30b du manganèse) sont également reportés. Sans additif, le transitoire obtenu en solution zinc-manganèse à 10 mAcm-2 (transitoire 30c) est parfaitement superposable à un transitoire d’une solution du chlorure de zinc (transitoire 30a). Dans ces conditions, la seule réaction qui se produit est la réduction des espèces zinc, le manganèse ne se dépose pas.
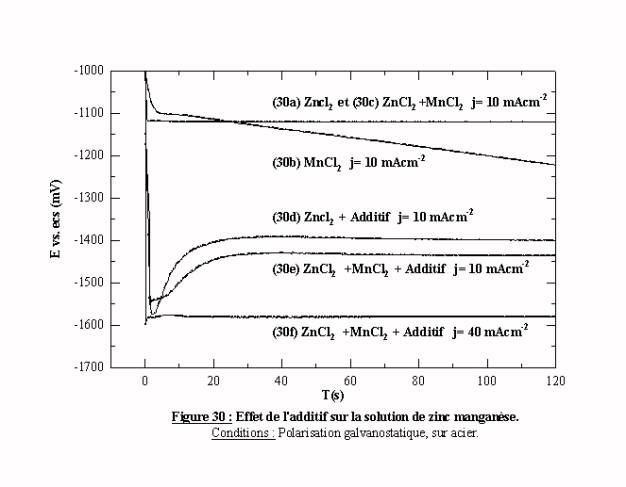
En présence d'additif (transitoire 30e), les transitoires sont semblables à ceux obtenus dans un bain de zinc avec additif (transitoire 30d). On note simplement une surtension supplémentaire induite par l'introduction du manganèse. Quand l'intensité du courant imposé est suffisante (supérieure à 20 mAcm-2), l'introduction de l'additif impose une stabilisation à des potentiels cathodiques par rapport au potentiel de réduction des espèces manganèse (transitoire 30f). Le déplacement des potentiels dans cette zone augmente la probabilité de déposer du manganèse.
III ) 4.4 Influence de la concentration en additif A (Base)
Jusqu'à présent, l'additif a été utilisé à une concentration nominale de 40 ml l-1. Cette concentration a été déterminée, par voltamétrie cyclique, en étudiant l'effet de la concentration d'additif sur les paramètres électrochimiques du système.
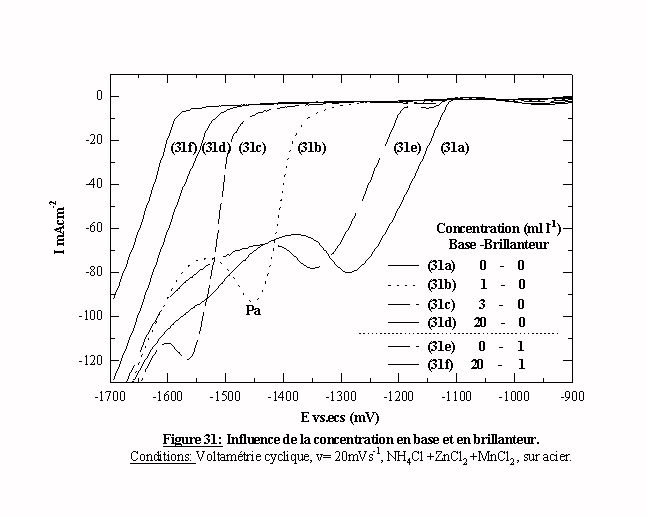
La figure 31 présente les balayages aller de l'électrolyte ZnCl2 + MnCl2 + NH4Cl avec 0, 1, 3, 20 ml l-1 d'additif. La courbe 31a est la courbe de référence correspondant au bain sans additif. Quand la concentration d'additif est égale à 1 ml l-1 (courbe 31b), le pic de cristallisation Pa se produit au potentiel E=-1330mV/ecs, avec une surtension de 300 mV par rapport à l’électrolyte sans additif (courbe 31a). Une faible quantité d'additif (base) induit alors une surtension importante. Quand on augmente la concentration d'additif jusqu'à une concentration égale à 3 ml l-1 (courbe 31c), le pic de cristallisation Pa se déplace vers les potentiels cathodiques. La surtension induite est ainsi fonction de la concentration en additif. On note également la conservation du palier de diffusion.
Pour les concentrations en additif supérieures à 7 ml l-1, le pic de cristallisation Pa débute au potentiel E=- 1500 mV/ecs. D'après ce résultat, la quantité nécessaire d'additif semble être de 7 ml l-1. Cependant, l’observation des dépôts montre une amélioration des dépôts entre 7 ml l-1 et de 40 ml l-1, plus la concentration d'additif est élevée, plus le dépôt est brillant et adhérant. Le critère "surtension" ne suffit donc pas à déterminer la concentration adéquate.
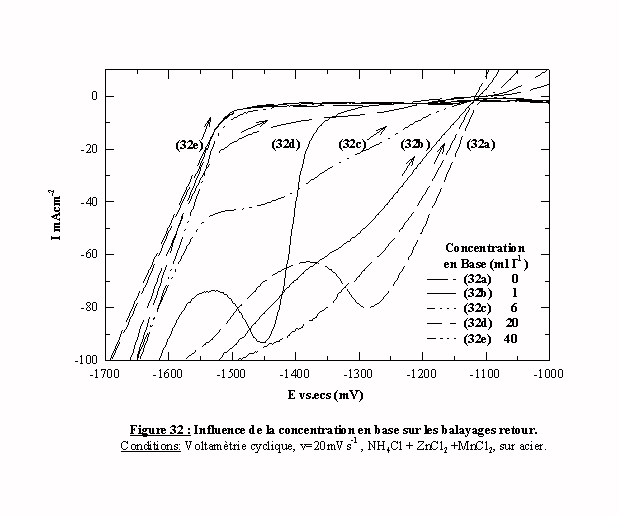
Les balayages retour subissent également l'influence de la quantité d'additif (figure 32). Quand la concentration en additif est égale à 1 mll-1 (courbe 32b), le balayage retour est très diffèrent du balayage aller. Quand la concentration en additif est égale à 40 mll-1 (courbe 32e), le balayage retour est identique au balayage aller. Les autres voltamogrammes nous donnent les positions intermédiaires entre ces deux états. Nous avons ainsi une relation entre la concentration d'additif et la relation entre les tracés aller et retour en voltamétrie cyclique. Cette relation pourrait être un critère utile à la détermination de la concentration adéquate d’un additif.
La figure 33 présente les parties ascendantes des pics anodiques. Quand la concentration en additif augmente, la vitesse de dissolution décroît sensiblement jusqu’à l’apparition d’un changement de pente très net (visible sur la courbe 33e obtenue avec 40 ml l-1).
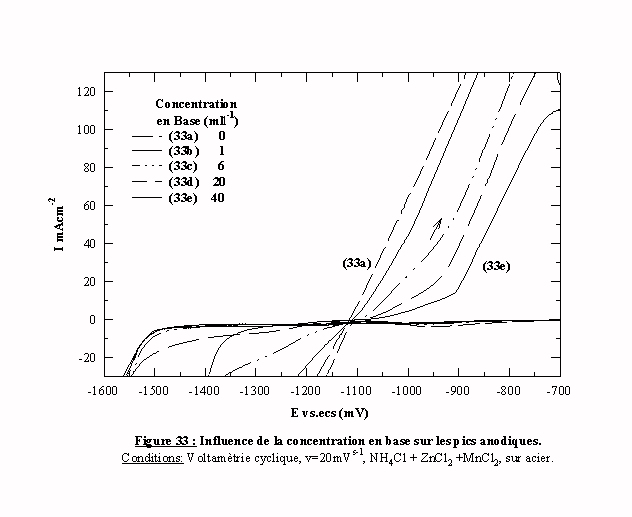
Nous pouvons en déduire que, plus la quantité d’additif est importante, plus la vitesse de dissolution est faible. Cependant, cet effet peut être dû, soit à la nature du dépôt lui même, soit à l'électrolyte dans lequel se fait la dissolution dans ce cas l'additif peut agir comme inhibiteur de corrosion. Afin d'évaluer la contribution respective de ces deux phénomènes, il faudrait effectuer la dissolution dans une solution commune avec une technique plus adaptée à la détermination des vitesses de dissolution.
III ) 4.5 Utilisation de brillanteur A
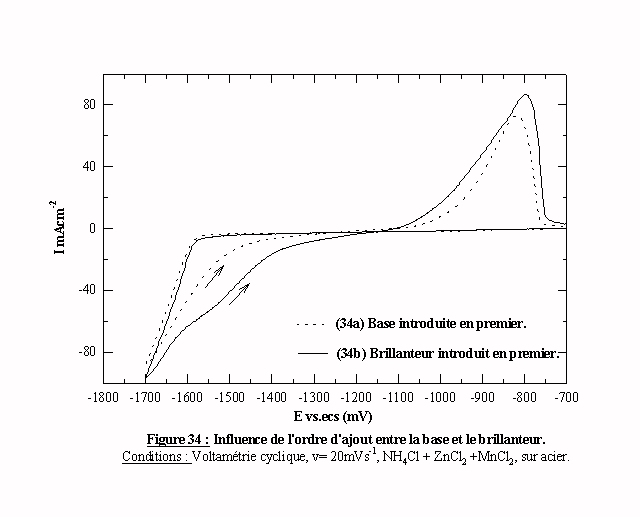
La majorité des additifs se présente sous la forme de deux produits séparés dont l'un est appelé base et l'autre brillanteur. L'étude précédente concerne la base. Le brillanteur produit les mêmes effets cinétiques que la base, mais son influence sur les paramètres électriques est différente. Les modifications induites par l'introduction du brillanteur sont reportées sur la figure 31. L’ajout de 1 ml l-1 de brillanteur dans une solution de ZnCl2 + MnCl2 + NH4Cl déplace le pic Pa de 30 mV (courbe 31e). Ce déplacement n’est pas comparable avec le déplacement de 300 mV induit par la même quantité de base. Quand on ajoute du brillanteur dans une solution contenant préalablement 20 ml l-1 de base, on déplace à nouveaux le pic Pa de 20 mV (courbe 31f), alors que l'ajout de base ne contribue plus à l'augmentation de la surtension induite. Le brillanteur a donc une action complémentaire de la base. L'emploi de ce brillanteur est d'autant plus intéressant qu'il augmente la surtension du système zinc-manganèse et la probabilité d'obtenir du manganèse dans le dépôt. Il reste à étudier s'il existe une influence de l’ordre d’ajout entre la base et le brillanteur. Sur la figure 34 nous avons présenté deux voltamogrammes obtenus dans deux électrolytes. L'électrolyte (34a) est composé: de ZnCl2 + MnCl2 + NH4Cl + 20 ml l-1 de base puis 1 ml l-1 de brillanteur. L'électrolyte (34b) est composé de ZnCl2 + MnCl2 + NH4Cl + 1 ml l-1 de brillanteur puis 20 ml l-1 de base. Les deux tracés présentent une différence sur le balayage retour. En effet pour l'électrolyte (34a) le retour est plus proche du balayage aller et le pic comporte une pente plus faible. De plus, quand on compare les dépôts obtenus avec ces deux solutions, le dépôt le plus brillant est celui élaboré à partir de l'électrolyte (34a). Donc l’ordre de préparation est important. D’après ces résultats la base sera toujours introduite avant le brillanteur.
III ) 5 Conclusion
Sans additif, la codéposition du zinc et du manganèse est normale. L'élément ayant le potentiel le moins cathodique (le zinc) se réduit au même potentiel que dans un bain ne contenant que l'espèce zinc. Pour réduire du manganèse, il faut se placer dans une zone de potentiel plus cathodique par rapport à –1520 mV/ecs. En régime potentiostatique, cette condition peut être remplie mais l'équipement industriel est classiquement configuré en mode galvanostatique. La mise au point d'un procédé d'élaboration doit prendre en compte cet aspect qui rend caduque l'exploration de cette voie. En régime galvanostatique, la valeur de potentiel nécessaire à la réduction des espèces manganèse ne peut être atteinte dans de bonnes conditions qu'en absence d'espèce zinc. Un potentiel suffisamment cathodique peut être atteint pour des densités de courant supérieures au courant limite car le zinc est placé sous contrôle cinétique de transfert de masse. Cependant, ces conditions d'élaboration conduisent à des dépôts brûlés ou poudreux avec des rendements très faibles (réduction massive d'hydrogène). L'obtention d'un dépôt d'alliage zinc-manganèse sans additif n'est pas possible.
L'introduction de l'additif dans le bain contenant le zinc et le manganèse modifie les paramètres cinétiques du système. L'aspect le plus intéressant des modifications est l'augmentation importante de la surtension de réduction des espèces zinc. Cet accroissement de surtension permet d'atteindre une zone de potentiel où la réduction des deux espèces électroactives est possible. L'additif impose un nouveau contrôle cinétique qui devrait permettre de préparer un alliage zinc-manganèse. Les conditions les plus intéressantes sont obtenues pour une concentration en additif de 40 mll-1 en base et 1 mll-1 en brillanteur. L'étude cinétique permet de dire que l'introduction de l'additif augmente la probabilité d'obtenir un dépôt de zinc-manganèse.
III ) 6 Etude de l'additif B
Les résultats obtenus au cours de l'étude cinétique permettent d'évaluer rapidement le comportement d'un additif. En effet, en fonction de sa capacité à induire une surtension à la réduction des espèces zinc, nous pouvons déterminer son aptitude à permettre la réduction des espèces manganèse. En suivant la même méthode que précédemment un autre additif nommé B est étudié.
III ) 6.1.1 Etude de l'influence de la concentration en base B
A partir du même bain contenant les chlorures zinc, de manganèse et d'ammonium, la figure 35 illustre l'évolution de l'allure des voltamogrammes aller en fonction de la concentration en base B. Les résultats sont conformes avec ceux obtenus en présence de l'additif A (cf. Figure 31). L'ajout de 1 mll-1 d'additif B induit une surtension de 210 mV (courbe 35b), puis la surtension induite devient proportionnelle à la concentration d'additif jusqu'à la valeur de 6 mll-1 (courbe 35a). Au delà de cette valeur jusqu'à 35 mll-1 tous les voltamogrammes ont la même allure que le voltamogramme (35e), pour lequel la surtension, pour cette concentration, est de 430 mV.

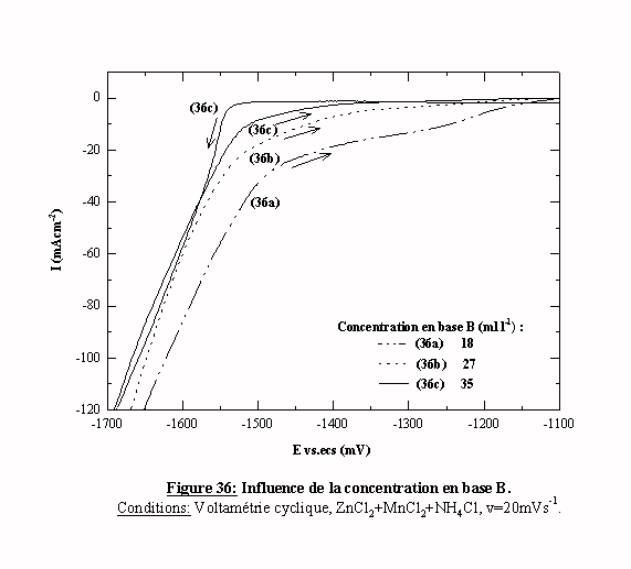
La figure 36 présente les balayages retour pour des concentrations de 18, 27, 35 mll-1. Les tracés obtenus quand la concentration est comprise entre 6 et 18 mll-1 prennent la même allure que le voltamogramme (36a) puis, proportionnellement à la concentration de base, le balayage retour se rapproche du balayage aller. Le tracé retour le plus proche du balayage aller est obtenu pour une concentration de 35 mll-1. Cette concentration est choisie pour la suite de l'étude et pour l'élaboration des dépôts.
III ) 6.1.2 Etude de la concentration en brillanteur B
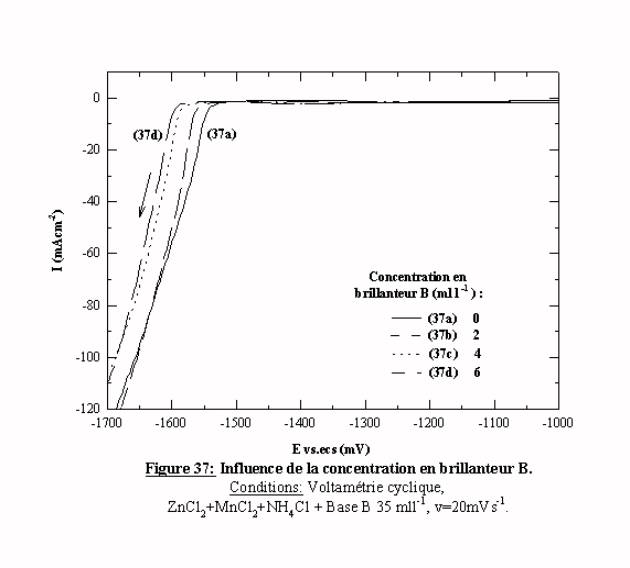
La figure 37 présente l'effet de la concentration en brillanteur sur l'électrolyte ZnCl2 + MnCl2 + NH4Cl + Base B (35 mll-1). Le brillanteur de cet additif adopte un comportement semblable à celui de l'additif A (cf. §III ) 4.5 et voltamogramme 31e): il ajoute une surtension complémentaire à celle induite par la base. Son effet sur la surtension est proportionnel à sa concentration jusqu'à la valeur de 6 mll-1 ou l'effet est maximum.
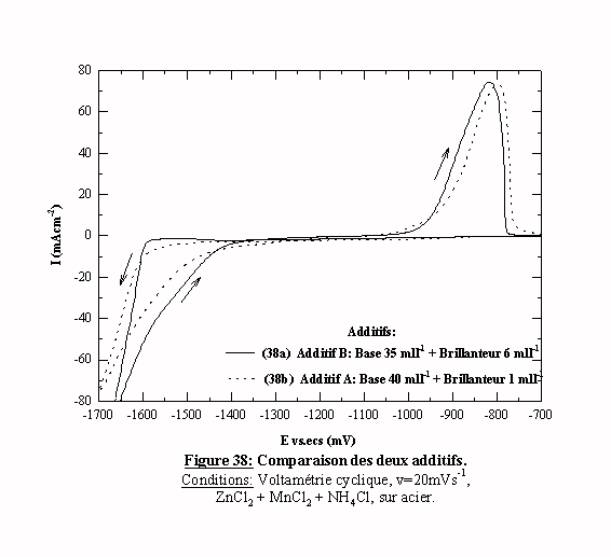
L'additif B se comporte d'une manière très similaire à celle de l'additif A, seules les valeurs relatives des surtensions induites varient. Pour apprécier cette différence, la figure 38 regroupe les voltamogrammes obtenus pour des concentrations optimales de base et de brillanteur, c'est à dire : 40 mll-1 de base et 1 mll-1 de brillanteur pour A et 35 mll-1 de base et 6 mll-1 de brillanteur pour B. Les deux voltamogrammes ont la même allure générale, les tracés des deux balayages sont très proches. Si on observe plus attentivement les deux balayages aller, on constate que les deux tracés se croisent. On peut définir ainsi deux domaines. Quand le potentiel est anodique par rapport à –1600 mV/ecs, l'intensité du tracé (38a) est inférieure à celle du tracé (38b), l'additif B exerce alors un blocage plus efficace que l'additif A. Dans cette zone de potentiel, en présence de A, le zinc est plus susceptible de se réduire au détriment du manganèse. Pour des potentiels cathodiques par rapport à –1660 mV/ecs, l'intensité enregistrée est supérieure pour l'additif A, cependant nous ne pouvons pas déterminer la proportion relative des espèces qui peuvent être réduites (Zinc, Manganèse, Hydrogène). Cette comparaison peut indiquer une meilleure possibilité de réduire le manganèse en présence de l'additif B par rapport à l'additif A.