III )
Etude cinétique
L’étude cinétique à partir d’une solution contenant plusieurs espèces électroactives est très complexe. Dans le cas du bain utilisé pour préparer les alliages zinc-manganèse, plusieurs réactions électrochimiques peuvent intervenir: la réduction des espèces zinc et manganèse et l’évolution de l’hydrogène. L’étude globale ne peut se faire qu’après une étude séparée des composants. Il n’est pas possible de séparer la réduction des espèces métalliques de la réaction d’évolution de l’hydrogène, par contre l’évolution de l’hydrogène, et l’influence de l’ajout d’additif sur cette réaction, peuvent s’étudier séparément et cela constitue une première approche de l’influence de l’additif sur substrat d’acier. Nous étudions successivement l’influence de l’additif sur l’évolution de l’hydrogène, c’est à dire à partir d’une solution ne contenant que le chlorure d’ammonium, puis les bains zinc et manganèse, et enfin le bain zinc plus manganèse. Ce chapitre concerne essentiellement l'étude cinétique détaillée de la base de l'additif A. Les résultats se rapportant au brillanteur A et l'additif B sont présentés respectivement dans les chapitres III) 4.5 et III) 6.
III ) 1 Influence de l’additif A (Base) sur l'évolution de l’hydrogène
Le premier constituant d'un bain est le sel de fond, dans ce cas, le chlorure d'ammonium. Ainsi nous pouvons caractériser la seule réaction possible en polarisation cathodique : l’évolution de l’hydrogène sur le substrat d'acier. Ce système constitue également un premier support d'étude sur le mode d'action de l'additif car il permet de déterminer si l'évolution de l'hydrogène est sensible à la présence de l'additif et d'autre part si l'additif est lui-même électroactif. Les méthodes électrochimiques : voltamétrie cyclique et polarisation galvanostatique dans le domaine cathodique, tracé des droites de Tafel en mode potentiostatique et mesure de capacités de double couche par impédance électrochimique en présence ou en l'absence d'additif sont utilisées.
Les résultats que nous présentons sont les plus significatifs, ils illustrent de manière simple les déductions issues de nombreux essais effectués dont la présentation exhaustive aurait rendu la lecture fastidieuse.
III ) 1.1 Etude en voltamétrie cyclique
La figure 1 présente les balayages aller des voltamétries cycliques effectuées sur l'acier dans le chlorure d'ammonium avec et sans additif. Le voltamogramme 1a, effectué à la vitesse de 20 mVs-1, sans additif, présente une augmentation régulière du courant en fonction de l'augmentation de la surtension. Dans ce milieu, la seule réaction cathodique possible est la réduction du proton dont le potentiel électrochimique théorique est EH+/H2 = - 600 mV/ecs au pH 5-6. La courbe montre que le courant cathodique n'augmente qu'à partir d'un potentiel proche de E1= -1000 mV/ecs, mettant en évidence la surtension de d'évolution de l'hydrogène sur acier. Si nous effectuons le même type de polarisation à la vitesse de 0,5 mVs-1 (voltamogramme 1b), la réponse enregistrée est très proche en intensité et en potentiel. Les résultats sont identiques pour des vitesses comprises entre 0,5 et 100 mVs-1. L'évolution de l'hydrogène dans le chlorure d'ammonium sur acier n’est pas fonction de la vitesse de balayage.

Le voltamogramme 1a', obtenu en présence d'additif à 20 mVs-1, conserve, pour de faibles surtensions, la même allure générale qu’en l’absence d’additif. L'absence de nouveau pic cathodique indique que l'additif ne contient pas d'élément susceptible d'être réduit dans la zone de potentiel étudiée. Néanmoins la présence d'additif provoque un changement de pente de la courbe potentiel-courant à partir du potentiel E=-1400 mV/ecs. Quand la vitesse de balayage est de 0,5 mVs-1 (Figure 1b'), l'intensité du courant mesurée est très inférieure à celle obtenue à 20 mVs-1. En effet pour une tension E=-1600 mV/ecs, l’intensité du courant de réduction est divisée par 5. L'influence de la vitesse de balayage et le changement de pente sur la courbe 1a' traduisent l'existence d'une composante cinétique dans le processus réactionnel.
III ) 1.2 Etude en polarisation galvanostatique
La figure 2 présente les transitoires potentiel-temps, obtenus pour des densités de courant cathodique de 5 et 60 mAcm-2, en présence et en absence d'additif. Les quatre transitoires présentent une décroissance rapide puis une stabilisation très lente. Pour chaque valeur d'intensité de courant, l'introduction de l'additif induit un déplacement cathodique du potentiel de stabilisation et donc induit une surtension à la réaction d'évolution de l'hydrogène. Cette surtension est de l’ordre de la centaine de millivolt.
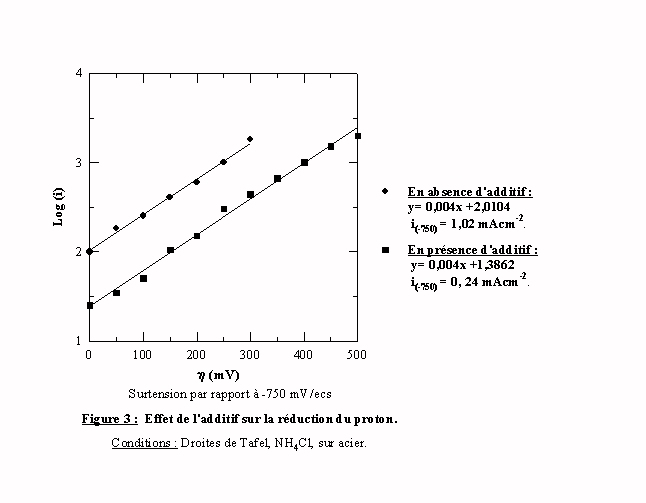
III ) 1.3 Droites de Tafel
Les voltamogrammes obtenus précédemment, montrent que le système évolue en fonction du temps, donc nous ne pouvons pas utiliser des résultats obtenus en régime dynamique, comme la voltamétrie cyclique, pour tracer les droites de Tafel. Elles ont été tracées en polarisation potentiostatique en notant pour chaque potentiel, la valeur du courant stationnaire. En présentant les résultats sous la forme Log(i) en fonction h et en utilisant la relation de Tafel concernant des valeurs de surtension suffisamment grandes, on détermine aisément le courant d’échange i0.
Les droites de Tafel obtenues avec et sans additif sont parallèles (Figure 3), ce qui indique que l’additif ne modifie pas le mécanisme de réduction en lui même, mais qu'il modifie la grandeur du courant d'échange. Ce courant d'échange est directement proportionnel à la surface électroactive. Nous pouvons émettre l'hypothèse que l'additif bloque une fraction de la surface et dans ce cas, nous pouvons appliquer la relation :
q = 1- Iadd/I
q, I et Iadd représentant respectivement le taux de recouvrement, les courants avec et sans additif. On trouve ainsi un taux de recouvrement de 75%.
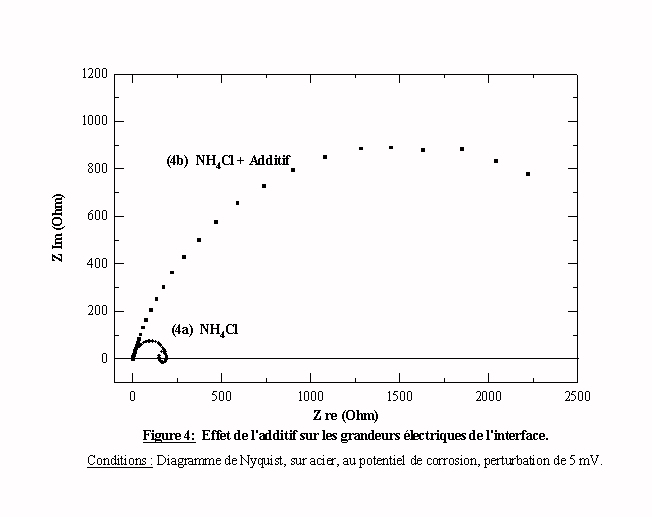
III ) 1.4 Diagramme d’impédance
Le but est d’exprimer les ordres de grandeur de la résistance de transfert et la capacité du système constitué d'une électrode d'acier plongée dans le chlorure d'ammonium avec et sans additif. Dans le chlorure d'ammonium, on obtient un diagramme (diagramme 4a) simple pouvant être modélisé par un circuit équivalent classique du type R(RQ) (cf. § II) 2.4). Ce diagramme comporte également une boucle inductive. Cette boucle inductive est classiquement attribuée à un phénomène de relaxation du courant avec le potentiel induit par la corrosion de l’acier [44]. En présence d’additif (diagramme 4b), on obtient également un diagramme classique type R(RQ) mais la boucle inductive disparaît. Cette disparition peut signifier que l’additif agit comme inhibiteur de corrosion. Excepté la boucle inductive, la comparaison des deux diagrammes ne met pas en évidence l’apparition d’un nouvel élément dans le circuit électrochimique équivalent. Par exemple, il n'apparaît pas d’autre capacité pouvant résulter d’un film actif adsorbé à l’interface. Donc l’additif n’agit pas en modifiant activement l’interface.
Comparons maintenant les grandeurs électriques (capacité de double couche et résistance de transfert) des deux diagrammes. Une première évaluation de la capacité est obtenue à l’aide de la fréquence mesurée au maximum du demi cercle par la relation wmaxRtC=1 avec w=2pf, les résistances sont déduites d’une lecture sur l’axe des réels. Une meilleure évaluation est également donnée par un ajustement algorithmique des paramètres à partir d’une modélisation d’un circuit équivalent (cf. § II) 2.4). La synthèse des résultats nous donne le tableau suivant.
|
Présence d’additif |
Potentiel mV/ecs |
Re Wcm2 |
Rt W cm2 |
Q w(n-1) mF cm-2 |
n |
C mF cm-2 |
Observation |
|
Non |
Ecorr=-697 |
1 |
192 |
877 |
0,82 |
592 |
Boucle inductive |
|
Oui |
Ecorr=-600 |
1 |
2650 |
172 |
0,78 |
94 |
|
L’analyse de ces données nous amène à faire plusieurs observations:
* La résistance de l'électrolyte reste constante, l’additif n’a pas d'effet sur la conductivité de la solution.
** L’additif augmente la résistance de transfert d’un ordre de grandeur et divise simultanément la valeur de la capacité.
La valeur d’une résistance électrique est inversement proportionnelle à l'aire réactive contrairement à la capacité qui est proportionnelle à l'aire réactive. Nous sommes donc en présence d'un effet qui résulte de la réduction de l'aire électroactive par adsorption à l'interface des molécules constitutives de l’additif. En effet, les molécules qui occupent des sites actifs, peuvent empêcher tout transfert électronique, c'est à dire réduire effectivement la surface électroactive. Ces résultats sont conformes à l'hypothèse concernant le blocage des sites actifs par l’additif.
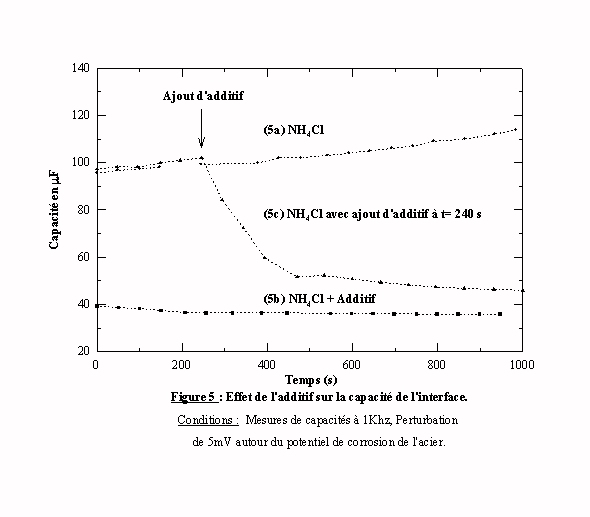
III ) 1.5 Mesure de la capacité en fonction du temps
Contrairement à l’hypothèse émise lors de la mesure précédente, le système ne reste pas à un état stationnaire mais évolue au cours du temps. Un suivi de la valeur de la capacité de double couche en fonction du temps peut se révéler intéressant. La technique utilisée durant ce paragraphe est décrite dans le chapitre Impédance électrochimique (cf. § II) 2.4)
Les valeurs des capacités qui sont données dans ce paragraphe ne peuvent être en aucun cas comparées à celles du paragraphe précédent. En effet, on assimile le circuit à un modèle uniquement capacitif, de plus chaque valeur est calculée à partir d'une mesure instantanée. Les grandeurs des capacités présentées dans ce paragraphe n'ont qu'une valeur relative. La figure 5 illustre l’évolution des capacités en fonction du temps de l’acier dans le chlorure d'ammonium avec et sans additif au potentiel de corrosion. Sans additif (courbe 5a), la capacité mesurée prend une valeur proche de 100 mF et augmente très légèrement au cours du temps. Cette augmentation résulte de la corrosion non uniforme de l’acier qui augmente l'aire réactive. En présence d’additif (courbe 5b), la capacité acquiert une valeur proche de 40 mF. Les valeurs des capacités étant suffisamment différentes en fonction de la présence ou de l’absence d’additif, nous avons un moyen de suivre, en fonction du temps, le processus d’adsorption. La courbe 5c, nous montre l’évolution de la capacité de l’acier dans le chlorure d'ammonium quand on ajoute l’additif après 240 secondes d’immersion. Plusieurs dizaines de minutes sont nécessaires pour retrouver la valeur de la capacité de la courbe 5b. Le fait d’avoir une diminution de la capacité au cours du temps indique que l’additif est capable d’occuper un site préalablement occupé par un proton. Ainsi l'additif présente une plus grande affinité avec la surface que le proton. Cet essai indique que le processus d'adsorption est l’enjeu d’une compétition entre le proton et la molécule d’additif pour chaque site et que la cinétique de l'adsorption est lente.
III ) 1.6 Conclusion
Les résultats obtenus montrent que l’additif agit en bloquant les sites électroactifs de la surface. La surtension constatée en voltamétrie cyclique et en polarisation galvanostatique s'explique par l'occupation et l'encombrement des sites favorables à la réduction de l'hydrogène atomique par les molécules d'additif adsorbées. Cet aspect peut expliquer la variation des réponses intensité-potentiel en fonction des conditions expérimentales car le proton et les molécules d’additifs sont soumis à une compétition pour occuper les sites actifs. Tous les essais effectués en régime dynamique (voltamétrie cyclique à grande vitesse) favorisent l'adsorption du proton donc sa réduction. Par contre, pour les essais effectués en régime stationnaire ou très lent (polarisation galvanostatique, courbe de Tafel, voltamétrie cyclique à faible vitesse), c’est l'adsorption des molécules de grandes tailles qui est favorisée contrairement à la réduction du proton.
L’additif ne modifie pas le mécanisme de réduction mais il impose sa cinétique d’adsorption et de désorption en modifiant l'aire électroactive.
III ) 2 Influence de l’additif A (Base) sur la réduction des espèces zinc
Nous connaissons maintenant les aspects principaux du mécanisme d'action de l'additif par rapport à l'évolution de l'hydrogène. Cette étude constitue une première approche pour étudier le comportement du même système en présence des cations zinc, principale composante métallique du dépôt. L'étude de l'effet de l'additif sur l'électrocristallisation du zinc utilise une méthodologie identique, bien que la présence des deux espèces électroactives enlève tout sens à certaines techniques comme l'établissement des droites de Tafel ou les mesures d’impédance.
III ) 2.1 Etude en voltamétrie cyclique de la réduction des espèces zinc
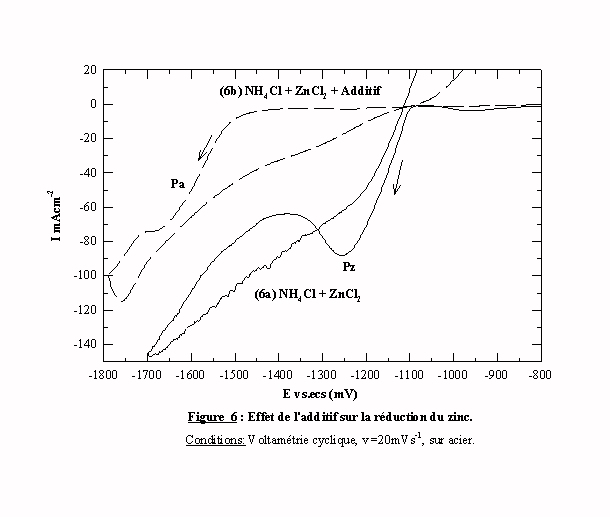
Cette partie est consacrée à la description et à la comparaison des voltamogrammes obtenus dans un bain contenant les espèces zinc. La figure 6 présente deux voltamogrammes obtenus à une vitesse de 20 mVs-1 respectivement sans additif (voltamogramme 6a) et en présence d'additif (voltamogramme 6b).
Le voltamogramme 6a présente un pic Pz de réduction au potentiel E=-1080 mV/ecs qui correspond à la formation du dépôt de zinc. En effet, on observe visuellement, lors du passage à ce potentiel, la formation d'un dépôt sur l'électrode. Ce pic de courant est suivi d'un palier diffusionnel mettant en évidence le contrôle cinétique par transfert de masse. La valeur de la densité de courant, à laquelle intervient ce palier, nous donne la valeur du courant limite. A partir du potentiel E= -1440 mV/ecs, se produit une augmentation du courant accompagnée d'un dégagement gazeux à la cathode. Le dépôt de zinc étant formé, le substrat d’acier n'est plus en contact avec la solution. L'augmentation du courant correspond donc à l'évolution de l'hydrogène sur le dépôt de zinc. Le balayage retour, plus accidenté est caractérisé par l'absence de croisement avec le balayage aller. Cette absence de boucle de germination montre que le dépôt métallique ne se produit pas directement par une germination tridimensionnelle (3D). La courbe retour est également marquée par une transition courant cathodique-anodique caractérisant une dissolution rapide du dépôt.
L'ajout de l’additif dans l'électrolyte ZnCl2 + NH4Cl change radicalement l'allure du voltamogramme (courbe 6b). Les étapes très précises et caractéristiques de l'électrocristallisation du zinc sont moins visibles. La partie cathodique présente deux parties distinctes: une partie à courant très faible et un pic de réduction Pa -1480 mV/ecs. En présence d'additif, le pic Pz de réduction des espèces zinc semble être déplacé vers des potentiels plus cathodiques. En effet, ce pic est très semblable au pic obtenu sans additif mais avec un décalage en potentiel de 400 mV. Dans cette zone de potentiel se produit un très faible dégagement gazeux correspondant à une légère évolution de l'hydrogène. Ce pic Pa concerne donc le dépôt de zinc sous influence de l'additif qui induit une surtension de cristallisation. L'inflexion observée sur le pic est probablement la conséquence de la réduction d'hydrogène. Le balayage retour est comparable à celui obtenu sans additif, mais avec une intensité de courant divisée par deux. Le passage à courant nul des deux balayages retour se fait au même potentiel E(i=0) =-1110 mV/ecs avec un changement de pente qui révèle une modification de la cinétique de la dissolution. Alors que le zinc seul présente les caractéristiques d'une dissolution rapide, le zinc en présence d'additif s'apparente plus à un système à dissolution lente ce qui indique que l’additif joue un rôle d’inhibiteur de corrosion.
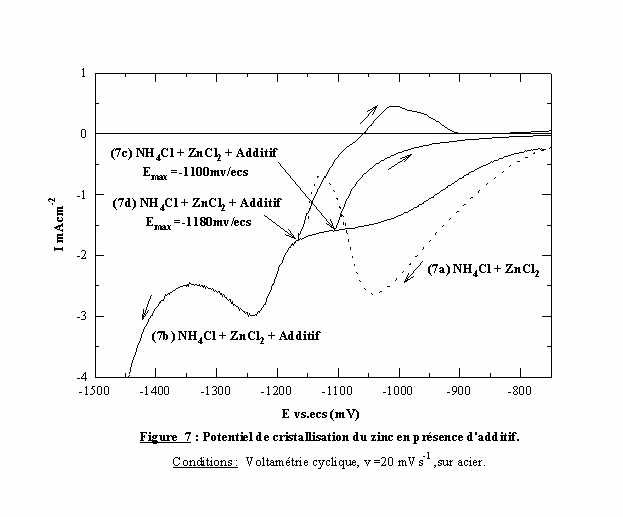
Le voltamogramme de la figure 6 est tracé sur une échelle de courant relativement importante et un agrandissement dans la zone de potentiel de -750 à -1500 mV/ecs, pour des courants inférieurs à 3,5 mA cm-2, est effectué pour détecter d'éventuels pics (Fig. 7). La courbe 7b montre que, dans cette zone de potentiel, le courant n'est pas nul et que deux pics apparaissent. On peut déjà noter que le pic à -1250 mV est dans la zone de potentiel du dépôt de zinc sans influence de l'additif.
Pour attribuer un processus électrochimique à ces deux pics, deux voltamogrammes sont tracés: l'un en changeant le sens de balayage en potentiel à -1100 mV/ecs, c'est à dire avant le pic (courbe 7c); l'autre à -1180 mV, c'est à dire au début du pic (courbe 7d). Le balayage retour de la courbe 7d passe au potentiel à courant nul pour la valeur correspondant au potentiel mixte du zinc dans ce bain, et montre ensuite un pic anodique correspondant à la dissolution du zinc. Le tracé retour de la courbe 7c ne montre pas de pic anodique, donc le premier pic ne correspond pas à un dépôt de zinc. Sur la même figure, un agrandissement de la courbe sans additif est aussi présenté (courbe 7a). Un pic à -1050 mV/ecs (pic ZH) précède le dépôt de zinc et ne correspond pas à un dépôt de zinc. Cette zone de potentiel, et ce premier pic ZH, sont étudiés au paragraphe suivant.
Le voltamogramme 7b montre, qu'en présence d'additif, le zinc se dépose en deux étapes: en faible quantité dans la zone de potentiel correspondant au dépôt sans additif et en dépôt massif dans la zone de potentiel plus cathodique.
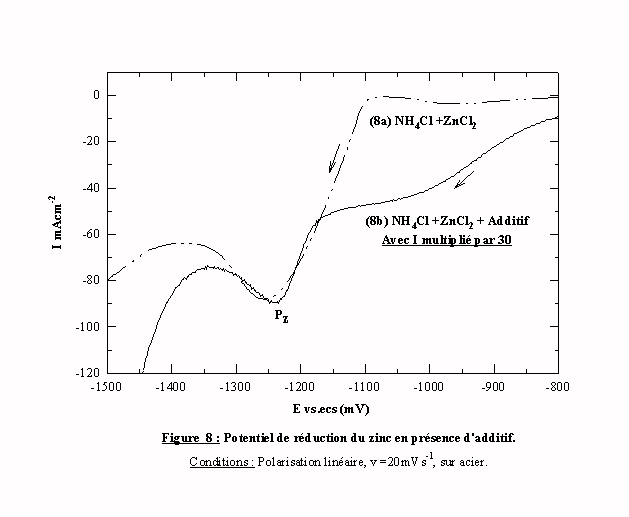
Pour confirmer ce résultat, nous avons superposé (Figure 8), le voltamogramme du bain sans additif (voltamogramme 8a) et le voltamogramme obtenu en présence d'additif en multipliant l'intensité de courant par 30 (voltamogramme 8b). La concordance est parfaite entre les pics Pz dans la zone de potentiel de réduction des espèces zinc. Nous avons montré dans le cas de la décharge du proton que le recouvrement de la surface est partiel. On peut ainsi proposer l'hypothèse de la présence de "trous" dans la couche d'additif dans lesquels le zinc peut se déposer sans subir son influence.

III ) 2.2 Etude de l'avant pic ZH
La figure 9 présente un agrandissement de la figure 6 dans la zone de potentiel précédant la réduction des espèces zinc. Le voltamogramme 9a est obtenu avec le bain sans additif. Cette figure met en évidence la présence d’un pic dont l'allure reste différente d'un pic de réduction classique. Ce type de pic a été également observé en milieu KCl [37]. Nous le désignons sous le terme d' "avant-pic" ZH . La partie initiale de l'avant-pic ZH est commune avec le voltamogramme 9d de la solution contenant uniquement le chlorure d'ammonium qui correspond à l’évolution de l’hydrogène sur l'acier. Donc il semble que le courant observé est dû dans les deux cas à l'évolution de l'hydrogène. En présence d'espèces zinc, (voltamogramme 9a) l'évolution de l'hydrogène serait ralentie puis bloquée. Ce blocage, situé dans une zone de potentiel anodique par rapport au potentiel d’équilibre du système zinc, est néanmoins imputable à l'introduction de cations Zn2+.
Cette zone correspond à la zone de dépôt en sous potentiel du zinc. Un mécanisme de déposition en sous potentiel (UPD) du zinc sur l’acier, l’argent, l’or et le platine, a déjà été démontré [45-49]. En effet, durant l'électrodéposition d’un métal sur un substrat différent, une monocouche peut être déposée dans une zone de potentiel plus anodique par rapport au potentiel d’équilibre. Des critères ont été établis pour prouver qu’un pic de voltamogramme est issu d’un processus d’UPD. Ces critères concernent :
![]() La présence d’un pic
anodique sur le balayage retour.
La présence d’un pic
anodique sur le balayage retour.
![]() L'annulation du courant
après l'arrêt du balayage en potentiel dans la zone correspondant à l’UPD,
indiquant un processus d’adsorption réversible.
L'annulation du courant
après l'arrêt du balayage en potentiel dans la zone correspondant à l’UPD,
indiquant un processus d’adsorption réversible.
![]() L'ordre de grandeur de
la charge sous le pic qui doit correspondre à la charge d’une monocouche (500mCcm-2).
L'ordre de grandeur de
la charge sous le pic qui doit correspondre à la charge d’une monocouche (500mCcm-2).
Le pic ZH est-il un pic de dépôt en sous potentiel de zinc (UPD) ?
Etudions l'hypothèse que cet avant-pic ZH soit un pic d'UPD de zinc. On effectue deux voltamogrammes avec des changements de sens de balayage aux potentiels respectifs de -1075 et -1100 mV/ecs (9b et 9c). Ceux-ci ne présentent pas de pic anodique, mais au contraire, des pics de réactivations cathodiques. De plus, en arrêtant le balayage durant la partie ascendante, l'intensité du courant ne s'annule pas. Enfin, la charge du pic est de l’ordre de grandeur de la dizaine de mC. Ainsi, ce pic ZH ne peut être considéré comme un pic d’UPD de zinc.
Cependant, il ne faut pas oublier que dans nos conditions expérimentales, la réduction de l’hydrogène et l’UPD de zinc se font dans la même zone de potentiel. De ce fait, la non justification de la présence de l'UPD par les trois critères devient caduque car la présence de cette réaction masque les observations que l'on fait quand l'UPD se produit seul [47].
Examinons l'hypothèse du blocage du dégagement d'hydrogène par l'UPD de zinc. Durant le balayage retour, la dissolution de l'UPD de zinc (courants anodiques) réactive la réduction de l'hydrogène (courants cathodiques). Le pic anodique devient alors indiscernable car seule la somme des courants est enregistrée. De même, la réaction de réduction d'hydrogène justifie l'absence d'annulation de l'intensité du courant et la charge sous le pic ZH.
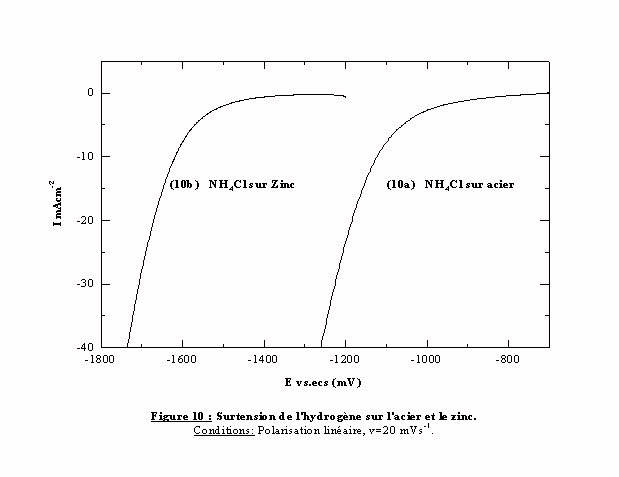
Le blocage de la réduction du proton dans une zone cathodique par rapport au potentiel de réduction des espèces zinc indique donc la présence d'un UPD de zinc. En effet, la formation de l'UPD de zinc augmente la surtension d'évolution de l'hydrogène car la surtension d'évolution de l'hydrogène sur le zinc est supérieure à celle qui se produit sur l'acier. La figure 10 montre qu'elle débute pour des potentiels cathodiques par rapport à -1400m V/ecs (cf. figure 10). En admettant que la couche résultant de l'UPD de zinc possède les mêmes propriétés que le dépôt de zinc massif, la réduction de l'hydrogène sur ce substrat devient impossible dans cette zone de potentiel. Le dégagement d'hydrogène devient alors inversement proportionnel au recouvrement de la surface par l'UPD de zinc. Cet aspect explique très bien la réactivation de la réaction de réduction de l'hydrogène lors du balayage retour. Une autre méthode permet de mettre en évidence un dépôt en sous potentiel, c'est l'utilisation de la cellule de Devanathan.
Perméation
de l'hydrogène
La réduction des espèces zinc est toujours accompagnée de la réaction d'évolution de l'hydrogène suivant la réaction globale:
2 H+ + 2 e- è H2
Cette relation s'effectue en plusieurs étapes parmi lesquelles figure la formation d'atome d'hydrogène adsorbé:
H+ + e- è Hads
La formation d'un dépôt en UPD étant aussi un processus d'adsorption, une compétition a lieu pour occuper les sites actifs sur le substrat d'acier. Dans le cas d'un substrat tel que l'acier, les atomes Hads peuvent être absorbés dans l'acier:
Hads è Habs
La quantité d'hydrogène absorbée dans l'acier dépend du taux de recouvrement q de l'acier par l'hydrogène adsorbé. L'absorption de l'hydrogène dans le substrat d'acier au cours de la formation du dépôt de zinc a des conséquences très importantes car elle conduit à la fragilisation de l'acier et à des risques de rupture. Le Laboratoire de Physico-Chimie des Matériaux s'est intéressé à ce problème [36,38], et a mis au point une cellule à perméation (cellule de Devanathan) pour permettre de déterminer la quantité d'hydrogène qui traverse la membrane d'acier en fonction des conditions de formation du dépôt de zinc.
Les atomes d'hydrogène qui, après perméation, se retrouvent de l'autre côté de la membrane, sont oxydés. Donc l'intensité du courant d'oxydation est reliée à la quantité d'hydrogène qui a traversé la cellule. Comme nous l'avons dit précédemment, il existe une relation entre le recouvrement de la surface par les atomes adsorbés et la quantité d'hydrogène absorbée. Si les espèces zinc se déposent en UPD, elles occupent des sites actifs et diminuent le nombre de sites disponibles, et de ce fait, la quantité d'hydrogène adsorbée ainsi que la quantité d'hydrogène qui traverse la membrane d'acier de la cellule. Donc la présence d'un dépôt de zinc en UPD doit diminuer la quantité d'hydrogène qui traverse la membrane d'acier.
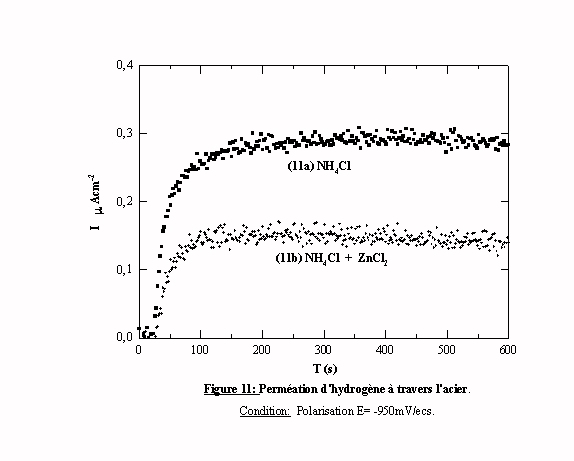
L'étude de l'évolution de l'hydrogène, dans la région d'UPD ne peut se faire qu'en mode potentiostatique car pour être sûr d'étudier l'UPD, il faut rester dans la zone de potentiel où elle intervient. Les essais de perméation ont été effectués à partir de deux solutions: la solution de NH4Cl qui ne peut conduire qu'à une adsorption de l'hydrogène et le bain de dépôt de zinc pour lequel les réactions liées à l'hydrogène et au zinc en UPD peuvent intervenir. La figure 11 présente les résultats d'essais de perméation effectués avec une polarisation à -950 mV/ecs de la cellule de chargement contenant une solution de chlorure d'ammonium avec (transitoire 11b) et sans zinc (transitoire 11a). Dans les deux cas les deux tracés présentent la même allure générale. Dès que la polarisation s'établit, le courant augmente rapidement jusqu'à l'établissement d'un courant stationnaire de perméation. Le chlorure d'ammonium donne une valeur de courant stationnaire proche de 0,30 mAcm-2, en présence de zinc cette valeur est proche de 0,15 mAcm-2. Cet essai met bien en évidence une compétition entre les espèces Hads et Zn(I) ads pour l'occupation des sites électroactifs et confirme les résultats obtenus en voltamétrie cyclique.
Le pic
ZH en présence d'additif
Le pic ZH n'est pas mis en évidence dans l'électrolyte contenant le zinc et l'additif (voltamogramme 7b). L'augmentation de courant se fait d'une manière régulière jusqu'au potentiel E=-1180 mV/ecs, sans formation de pic, c'est à dire sans blocage du processus d'évolution de l'hydrogène comme dans le cas de la solution sans additif. Cette valeur du courant reste nettement inférieure au voltamogramme 7a ce qui montre que l'additif inhibe à la fois la réduction du proton et l'UPD de zinc. En effet, les sites électroactifs de l'interface sont l'enjeu d'une compétition d'adsorption entre les trois espèces contenues dans l'électrolyte (Hads, Zinc en UPD, additif). Dans un premier temps, la présence d'additif inhibe la réduction de l'hydrogène en réduisant le nombre de sites actifs, conformément aux résultats du paragraphe III ) 1.6 . Dans un deuxième temps, l'adsorption des espèces zinc en sous potentiel, se trouve défavorisée car elle n'intervient qu'à partir d'une certaine valeur de potentiel sur une surface préalablement occupée par l'hydrogène et l'additif. La contribution de ces deux phénomènes conduit à la disparition du pic ZH en présence d'additif.
III ) 2.3 Etude de l'influence de la vitesse de balayage en voltamétrie cyclique
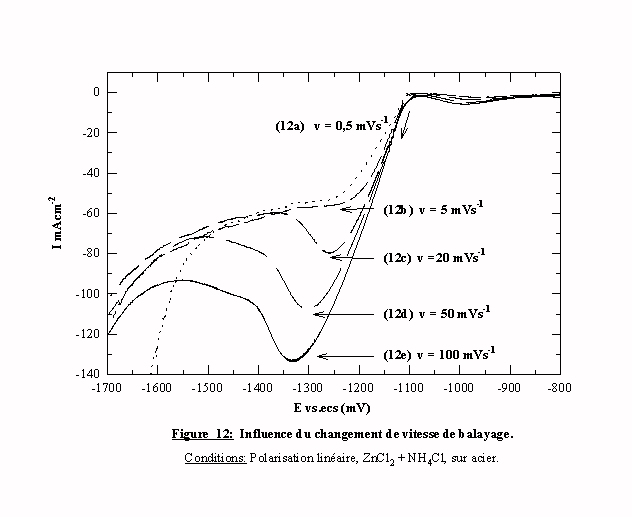
Les voltamogrammes présentés précédemment sont tracés à la vitesse de 20 mVs-1. Or, nous avons montré que l'additif introduit une composante cinétique dans le processus de réduction du proton (cf. § III ) 1.1 ). Donc l'étude de l'influence de la vitesse de balayage sur l'allure des voltamogrammes obtenus dans des bains, en présence et en absence d'additif, devient nécessaire. La figure 12 regroupe les courbes tracées à partir d'une solution sans additif pour différentes vitesses de balayages. Quelle que soit la vitesse 0,5 ; 5 ; 20 ; 50 ou 100 mVs-1, les courbes obtenues gardent la même allure générale. On retrouve toutes les étapes caractéristiques de la réduction des espèces zinc avec une variation de l’intensité du pic de réduction qui intervient toujours au même potentiel. Ce paramètre est donc indépendant de tout facteur cinétique.
La même série d'essais est entreprise en présence d’additif (figure 13). L'évolution de l'allure des pics PZ concernant la réduction des espèces zinc sans influence de l'additif, semble identique à celle constatée sur la figure 12. Cependant, bien que la réduction des espèces zinc sans additif se produise toujours au même potentiel (cf. § III ) 2.1 ), il est difficile de mettre en évidence cet aspect sur les voltamogrammes à cause de la valeur faible des courants de ces pics. Par contre, on observe un déplacement du pic de réduction Pa des espèces zinc sous influence de l'additif de l'ordre de la centaine de millivolt en fonction de la vitesse de balayage. La surtension induite par l’additif est un paramètre dépendant d’une constante cinétique globale. Cette constante cinétique est le résultat d’une compétition entre des espèces contenues dans l’électrolyte pour l’occupation des sites électroactifs (cf. § III ) 1.6). Les constantes d’adsorption du proton, des espèces zinc et des molécules d’additif sont différentes et c’est le choix de la vitesse de balayage qui privilégie une adsorption plutôt qu’une autre.
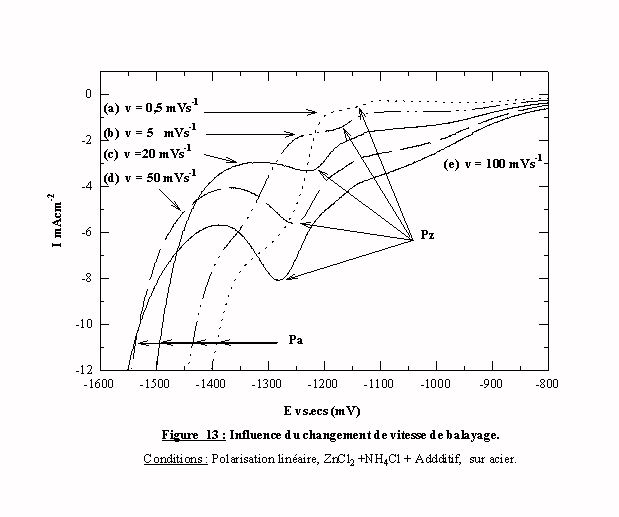
D'après la figure 13, quand le balayage est lent, la surtension induite est relativement faible donc la réduction des espèces zinc est privilégiée par rapport à l’adsorption de l’additif. Le résultat est inverse pour les balayages rapides. La constante d’adsorption de l'additif est donc inférieure à la constante d’adsorption des espèces zinc. Les résultats obtenus durant cette partie peuvent être groupés dans le tableau suivant:
|
Potentiel en mV/ecs |
ZnCl2 + NH4Cl /acier |
ZnCl2 + NH4Cl + Additif
/acier |
|
-750 < E < -950 |
Réduction de H+ |
Blocage partiel de la réduction de H+ |
|
-950 < E <-1080 |
UPD de zinc |
Blocage de l'UPD de Zinc |
|
-1080 < E < -1350 |
Réduction de Zn2+ |
Réduction très minoritaire de Zn2+ |
|
-1350 < E < -1500 |
Réduction de Zn2+ |
Réduction du Zn2+sous influence de l'additif |
|
-1550< E < -1700 |
Réduction de Zn2+ et H+ |
Réduction de Zn2+et H+ sous influence de l'additif |
|
Contrôle cinétique |
Transfert de masse |
Adsorption de l'additif |
|
Mode de croissance |
Pas de germination 3D |
|
|
Pic anodique |
Dissolution rapide |
Inhibition de la dissolution |
III ) 2.4 Etude en polarisation potentiostatique
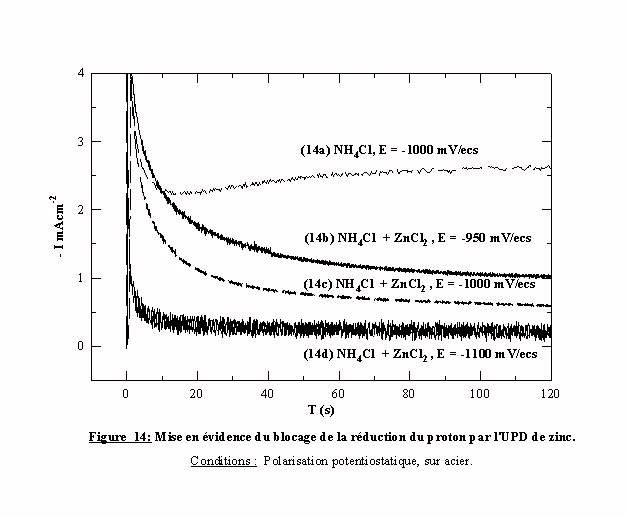
La figure 14 présente les transitoires Intensité-temps obtenus dans le chlorure d'ammonium en présence (transitoires (14b)(14c)(14d)) et en absence de zinc (transitoires (14a)), aux potentiels respectifs de -950,-1000,-1100 et -1000 mV/ecs, c'est à dire dans la zone de potentiel de l'avant pic. La comparaison des deux transitoires (14c) et (14a) avec et sans zinc à -1000 mV/ecs met en évidence le blocage de l'évolution de l'hydrogène par l'introduction du zinc. De plus, les transitoires (14b)(14c)(14d) montrent que l'intensité de stabilisation varie inversement à la surtension imposée. Plus le potentiel est proche du potentiel de cristallisation du zinc, plus la réduction de l'hydrogène est bloquée par l'UPD de zinc. Ces deux observations faites à l'état stationnaire confirment les hypothèses émises lors de l'étude menée en voltamétrie cyclique (cf. § III ) 2.1 ).
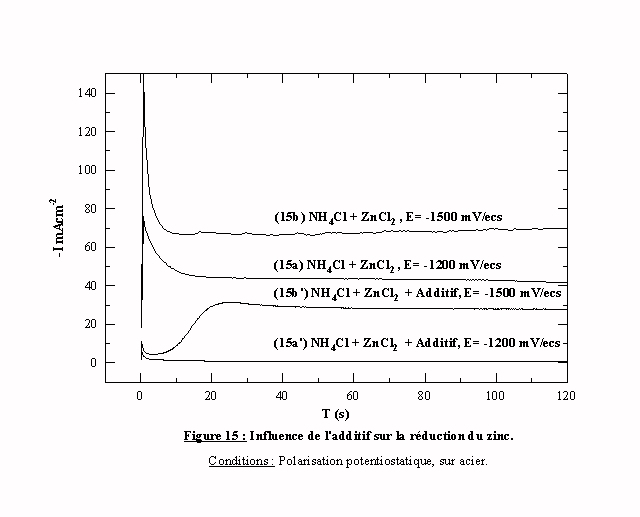
Quand le potentiel imposé est cathodique par rapport au potentiel de réduction des espèces zinc (figure 15, courbes (15a) –1200 mV/ecs et (15b) –1500 mV/ecs), les valeurs des intensités mesurées augmentent avec les surtensions imposées. Ces transitoires correspondent à la réduction des espèces zinc.

En présence d'additif, pour E=-1200 mV/ecs, on observe simplement une très forte diminution de l'intensité des courants de réduction. Celle-ci passe de 42 mAcm-2 (transitoire 15a), à 1 mAcm -2 (transitoire 15a’). Pour cette valeur de potentiel, l'additif bloque très fortement la réduction des espèces zinc.
Pour un potentiel imposé de -1500 mV/ecs en présence d'additif, le transitoire 15b' présente une allure différente. Durant les cinq premières secondes, le courant prend une valeur très faible puis augmente pendant vingt secondes jusqu'à une valeur de courant de 28 mAcm-2. Cette valeur reste cependant inférieure à moins de la moitié de celle obtenue sans additif. La valeur de l'intensité du courant de stabilisation montre une diminution de l'effet de blocage de la réduction des espèces zinc par l'additif à partir d'une certaine valeur de potentiel appliqué. A partir d'une certaine valeur de potentiel, l'intensité d'inhibition est inversement proportionnelle à la surtension cathodique. L'allure du transitoire indique également une augmentation du courant de réduction en fonction du temps. Cette évolution est liée à l'évolution de l'intensité du blocage induit par la désorption progressive des molécules d'additifs.
Sans additif, les essais menés en polarisation potentiostatique confirment le blocage de la réduction du proton par l'UPD de zinc. L'additif bloque totalement puis partiellement, en fonction du potentiel imposé, la réduction des espèces zinc et de l'hydrogène.
III ) 2.5 Etude en polarisation galvanostatique
Les transitoires, représentatifs des essais effectués, en mode galvanostatique, dans le bain de zinc sont présentés sur la figure 16. On observe deux types de transitoires en fonction de l'intensité de courant appliquée. Le transitoire 16a est représentatif des résultats obtenus pour des intensités de courant inférieures au courant limite. Son allure présente une décroissance puis une stabilisation à un potentiel proche de E1=-1230mV/ecs, caractéristique de la réduction des espèces zinc.
Quand l'intensité appliquée devient supérieure au courant limite, on obtient un transitoire du type 16b. Ce transitoire présente des oscillations entre deux potentiels E1 =-1230 mV/ecs et E2 =-1600 mV/ecs. Ce potentiel E2 correspond au potentiel de réduction de l'hydrogène sur le zinc comme le montre le transitoire 16c, tracé à partir d'un bain ne contenant que le chlorure d'ammonium sur un substrat de zinc pour une intensité de courant de 20 mAcm-2. La valeur du potentiel enregistré prend alternativement la valeur du potentiel de la réduction des espèces zinc puis le potentiel de la réduction du proton sur l'électrode. Ces oscillations sont en fait la conséquence du contrôle cinétique par transfert de masse de la réaction de réduction des espèces zinc. Quand l'intensité du courant est supérieure au courant limite, la couche diffuse est épuisée en espèces zinc car le flux des espèces électroactives contrôle la cinétique. Dans ce cas, la réduction de Zn2+ ne peut plus se faire et la réduction de proton se produit. Les bulles d'hydrogène formées produisent, en se dégageant, une agitation de la solution qui diminue l'épaisseur de la couche diffuse et permettent de réalimenter la proximité de la surface en espèces zinc. Pour confirmer cette hypothèse, nous avons fait intervenir une agitation mécanique durant une polarisation avec un courant supérieur au courant limite. Dès le début de l'agitation, on observe la disparition des oscillations et la stabilisation du système au potentiel E=-1230 mV/ecs comme pour la courbe (16a), car la modification des conditions hydrodynamiques provoque une diminution de l'épaisseur de la couche diffuse et donc une augmentation du flux des espèces à la surface. La réduction des espèces zinc, à la surface de l'électrode, étant toujours possible, la valeur du potentiel ne varie plus. Ainsi, en l'absence d'agitation, les deux réactions se produisent alternativement en fonction de la présence ou non d'espèces zinc. Une telle alternance de ces deux réactions implique la présence de bulles d'hydrogène piégées dans le dépôt ce qui confirme que la préparation des dépôts doit se faire avec une densité de courant inférieure au courant limite [37].
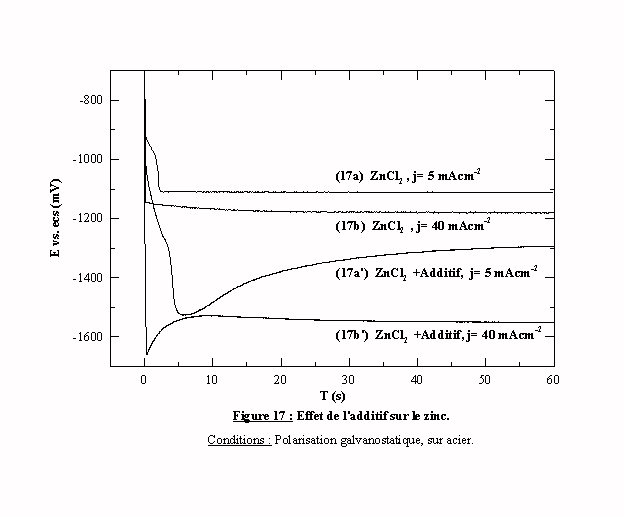
La figure 17 présente les transitoires d'une solution de chlorure d'ammonium et chlorure de zinc en présence et en absence d'additif pour des densités de courant inférieures au courant limite de 5 et 40 mAcm-2 sur l'électrode d'acier. Après un délai inhérent à la cinétique d'adsorption de l'additif à la surface, nous remarquons que les électrodes plongées dans les électrolytes qui contiennent l'additif prennent des potentiels nettement plus cathodiques qu'en son absence. Pour une intensité de courant imposé constante, l'ajout d'additif permet de déplacer cathodiquement le potentiel de réduction du système. Cet aspect est l'un des plus intéressants, car la plupart des dépôts sont effectués en mode galvanostatique et l'introduction d'une telle surtension peut contribuer à obtenir une codéposition avec un élément ayant un potentiel de réduction plus cathodique.

III ) 2.6 Conclusion
L'électrocristallisation du zinc sur substrat d'acier est précédée par l'évolution de l'hydrogène sur l'acier. La formation d'un dépôt de zinc en UPD modifie le substrat, donc les processus de transfert de charge s'effectuent sur un substrat ayant les caractéristiques du zinc. Ce dépôt en UPD permet d'expliquer deux caractéristiques des voltamogrammes:
![]() L'arrêt de l'évolution
de l'hydrogène (pic ZH).
L'arrêt de l'évolution
de l'hydrogène (pic ZH).
La surtension d'évolution de l'hydrogène sur zinc étant beaucoup plus importante que sur l'acier, dès que le dépôt de zinc en UPD est formé, l'évolution de l'hydrogène s'arrête car la zone de potentiel du pic ZH ne permet pas d'effectuer la réduction du proton sur le zinc.
![]() L'absence de germination
3D.
L'absence de germination
3D.
Il est admis que sur un substrat qui n'est pas de la même nature chimique que le dépôt, l'électrocristallisation se produit par un mécanisme de germination et croissance tri-dimensionnelle. La présence d'un dépôt de zinc en UPD modifie la nature du substrat et le dépôt massif de zinc s'effectue sur un substrat de zinc, ne nécessitant plus de germination.
En présence d'additif, le dépôt de zinc massif sous influence de l'additif se produit avec une surtension cathodique par rapport à un dépôt de zinc sans additif. Ce dépôt massif est précédé par la formation d'un dépôt de zinc qui ne subit pas l'influence de l'additif.