II )
Dispositifs
expérimentaux et
méthodes expérimentales
II ) 1Dispositifs expérimentaux
Différents types de dispositifs expérimentaux électrochimiques sont utilisés en fonction de l'étude à effectuer:
![]() Pour l'étude cinétique:
une cellule à double compartiment.
Pour l'étude cinétique:
une cellule à double compartiment.
![]() Pour l'élaboration des
dépôts: une étude préliminaire en cellule de Hull, puis élaboration des dépôts
dans une cuve de 30 litres.
Pour l'élaboration des
dépôts: une étude préliminaire en cellule de Hull, puis élaboration des dépôts
dans une cuve de 30 litres.
![]() Pour la perméation
d'hydrogène: une cellule de Devanathan.
Pour la perméation
d'hydrogène: une cellule de Devanathan.
Cette partie est consacrée à la description de ces dispositifs expérimentaux.
II ) 1.1 Cellule électrochimique à double compartiment
Elle est constituée de deux compartiments de 150 cm3 en plexiglass séparés par un diaphragme poreux. La séparation des compartiments catholytique et anolytique est justifiée par la production de produits secondaires à l'anode (cf. I)2.2). La cellule est munie d'un support conçu pour fixer les trois électrodes nécessaires à la constitution d'une cellule électrochimique.
Electrodes de travail
Les réactions électrochimiques se produisent à l'interface de l'électrode de travail. La nature chimique et l'état de surface de ces électrodes conditionnent ces réactions.
L'essentiel de l'étude cinétique s'effectue sur un substrat d'acier 35C4 dont la composition chimique est donnée dans le tableau ci-dessous :
|
Acier 35C4 |
|
|
Elément d'alliage |
Teneur |
|
Carbone |
0,3 à 0,35% |
|
Manganèse |
0,6 à 0,9 % |
|
Silicium |
0,4 % |
|
Chrome |
0,7 à 1,1 % |
Des études complémentaires portent sur un substrat de carbone vitreux (référence TOKAI) dont la structure amorphe conduit à des dépôts formés par un mécanisme de germination tridimensionnelle.
Les électrodes d'acier ou de carbone vitreux sont montées sur un support constitué par une canne de verre coudée et une enveloppe de résine polymérisable à froid et inerte chimiquement. Le contact électrique est assuré par un fil de cuivre soudé ou collé par une laque d'argent. Les surfaces des électrodes d'acier et de carbone sont respectivement de 1,5 et 0,07 cm2. L'électrode de travail peut être représentée par le schéma suivant:

Montage d'une électrode.
Ce montage permet un polissage performant et rapide sur une polisseuse automatique (BUEHLER Motopol 8). Le polissage automatique assure la planéité de l'échantillon et la régularité de l'état de surface. Les disques de polissages utilisés sont à base de carbure de silicium, la lubrification est assurée par un jet d'eau. Le premier polissage suit un protocole défini dans le tableau suivant :
|
Polissage |
Type de disque |
Taille des grains (Norme FEPA) |
Vitesse de rotation (Tours/mn) |
Durée (Minutes) |
|
Grossier |
P240 |
50 mm |
240 |
2 |
|
Fin |
P600 |
30 mm |
200 |
4 |
|
Très fin |
P1200 |
15 mm |
200 |
7 |
La force appliquée à chaque échantillon est de 10 gcm-2, le sens de rotation de la platine support est contraire au sens de rotation des disques. Dans le cas des électrodes de carbone vitreux un dernier polissage est effectué sur un feutre doux enduit de pâte diamantée.
Avant chaque expérience, l'électrode d'acier est repolie sur un disque P1200 pendant une minute, puis plongée dans une cuve à ultrasons et rincée afin d'éliminer les particules provenant du polissage. L'électrode de carbone subit le même protocole avec un polissage sur feutre doux.
Contre électrode, électrode de référence, appareillage
La contre électrode permet la mesure et le contrôle de l'intensité de courant qui passe dans la cellule électrochimique. Elle est constituée d'une grille de platine, inerte dans ce milieu et disposée parallèlement à l'électrode de travail afin d'obtenir une homogénéité du champ électrique.
L'électrode de référence est une électrode au calomel, saturée en KCl (ecs) qui permet de mesurer le potentiel de l'électrode de travail. A titre indicatif, le potentiel standard de cette électrode, à 25°C, par rapport à l'électrode normale à hydrogène est de 241 mV. Dans ce travail, toutes les valeurs des potentiels sont exprimées par rapport à cette électrode de référence.
Les électrodes sont reliées à un potentiostat-galvanostat (EG&G PAR 273) interfacé avec un micro ordinateur. Le potentiostat-galvanostat impose une perturbation en courant (en potentiel) et mesure la réponse en potentiel (en courant). Le micro ordinateur permet de piloter le potentiostat-galvanostat, de collecter et traiter les données.
Choix des l'électrolytes
La présence d'ions ammonium est nécessaire pour obtenir un dépôt de manganèse (cf. § I)2.2 ), pour cela la base du bain est le chlorure d'ammonium. Les concentrations en chlorure de zinc et chlorure de manganèse sont élevées afin d'éviter un appauvrissement trop rapide de la solution en espèces électroactives.
Les électrolytes d'étude sont :
![]() une solution de chlorure
d'ammonium:
une solution de chlorure
d'ammonium:
NH4Cl (5 M),
![]() une solution de chlorure
d'ammonium et de chlorure de zinc:
une solution de chlorure
d'ammonium et de chlorure de zinc:
NH4Cl (5 M) + ZnCl2
(0,5 M),
![]() une solution de chlorure
d'ammonium et de chlorure de manganèse:
une solution de chlorure
d'ammonium et de chlorure de manganèse:
NH4Cl (5 M) + MnCl2
(0,4 M),
![]() une solution de chlorure
d'ammonium, de chlorures de zinc et de manganèse:
une solution de chlorure
d'ammonium, de chlorures de zinc et de manganèse:
NH4Cl (5 M) + ZnCl2 (0,5 M) + MnCl2 (0,4 M).
L'étude de l'influence de l'additif est menée sur ces bains. Nous utilisons un additif commercial identifié sous le nom de A. La formulation de cet additif est complexe et soumise au secret industriel. Nous pouvons simplement dire que l'additif contient des composés aromatiques sulfonés, des acides organiques mono ou polycarboxyliques, des composés hétérocycliques… . Lors de l'étude cinétique seule une des deux composantes de l'additif, la base, est étudiée. En effet, l'autre composante, le brillanteur, présente le même type de comportement cinétique (cf. figure 31). La concentration nominale en base est déterminée et fixée pour tous les électrolytes à 40 ml l-1 à la suite de l'étude menée lors du paragraphe III)4.4.
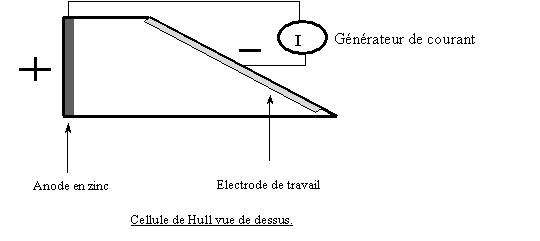
II ) 1.2 La cellule de Hull
Avant de commencer l'élaboration des dépôts, il est préférable d'effectuer des essais préliminaires. L'utilisation de la cellule de Hull permet de définir en quelques essais le comportement d'un bain sur une gamme d'intensité étendue. Le dispositif expérimental est composé de la cellule de Hull (300 cm-3) munie d'une contre électrode en zinc.

Les intensités locales appliquées sont fonction de l'intensité totale et de la distance à la contre électrode. La géométrie spécifique de la cellule de Hull utilise astucieusement cette propriété car une lecture directe sur des abaques permet de déterminer la distribution des intensités locales. Une agitation est produite par bullage d'air et par rotation d'un barreau magnétique. Le courant est fourni par un générateur qui délivre une intensité maximale de 10A.
L'électrode à revêtir est une tôle d'acier doux de 170 cm2 préparée selon le protocole suivant :
-Décapage électrolytique dans une solution soude pendant 5 minutes avec une intensité cathodique de 100 mAcm-2.
- Rinçage à l'eau.
- Décapage dans une solution d'acide chlorhydrique diluée à 50% avec un inhibiteur de corrosion (Hexaméthylènetetramyle).
- Rinçage à l'eau.
II ) 1.3 Cuve de 30 litres
Dans les conditions expérimentales du laboratoire, nous pouvons connaître et maîtriser les paramètres nécessaires à l'obtention de dépôt de zinc-manganèse et des dépôts ont été produits dans une cellule identique à celle où a été menée l'étude cinétique. Cependant, nous ne pouvons pas reproduire les conditions d'élaboration en laboratoire à une échelle industrielle car les paramètres physiques (intensité, potentiel, température, conditions hydrodynamiques ...) et chimiques (concentrations et pureté des espèces électroactives et des additifs, pH ...) sont forcément plus difficiles à maîtriser. De plus on ne peut pas utiliser une cellule à compartiment séparé. Pour ces raisons, une étude est menée afin d'élaborer et de caractériser des dépôts dans des conditions plus proches de la réalité industrielle.
Pour l'élaboration des dépôts nous disposons d'une cuve de 30 litres et d'un redresseur de courant. Le volume du bain étant conséquent (20 litres), il est donc nécessaire de filtrer et de produire une agitation par bullage d'air pour homogénéiser le bain. Le système est également régulé thermiquement à 25°C par une canne de chauffage couplée à un thermostat. Deux plaques de zinc font office d'anodes. Elles sont disposées à la même distance d'une part et d'autre de la tôle à revêtir. Ce dispositif ne permet pas de mesurer le potentiel de la réaction de réduction durant le dépôt. Or la maîtrise de ce paramètre est déterminante car le manganèse ne peut se déposer à partir –1600 mV/ecs (cf. § III)4.4) Nous allons donc considérer que nous appliquons une intensité telle que ce seuil soit dépassé.
II
) 1.4
La cellule de Devanathan
L'étude de la perméation de l'hydrogène à travers un acier lors de la formation d'un dépôt électrolytique peut se faire à l'aide de la cellule de Devanathan, mise au point par Devanathan et Starchurski [35],qui permet de mesurer la quantité d'hydrogène absorbée dans un métal lorsque celui-ci se trouve en situation cathodique de chargement. Une plaque métallique sépare la cellule en deux compartiments: un compartiment de chargement (face cathodique de la membrane) et un compartiment de détection (face anodique). Cette cellule est utilisée généralement pour déterminer le coefficient de diffusion de l'hydrogène à travers une membrane métallique. Pour notre part, nous ne nous sommes pas intéressés au coefficient de diffusion, mais nous avons utilisé cette technique pour déterminer la quantité d'hydrogène qui traverse une membrane d'acier en fonction des conditions d'électrolyse de la cellule d'entrée. La cellule utilisée, mise au point au laboratoire [36-38], utilise une plaque d'acier dont l'épaisseur moyenne est de 0,10 mm. L'étanchéité des compartiments est assurée par des joints toriques, un fil de laiton soudé à la plaque assure le contact électrique. La surface réactive de la plaque est de 10 cm2. Le protocole opératoire de préparation des plaques avant le montage est le suivant:
* rinçage à l'eau bipermutée sous ultra-sons,
* rinçage à l'aide d'une solution HCl à 10 % pendant 2 minutes,
* rinçage à l'eau bipermutée pendant 15 secondes.
Le bain à étudier est placé dans le compartiment de chargement. Le compartiment de détection contient une solution de soude décimolaire (NaOH 0,1 M) préparée à partir d'eau bipermutée et désaérée par bullage d'argon à température ambiante. Sur la face de sortie, un potentiel anodique par rapport au potentiel d'équilibre de l'hydrogène est appliqué pour oxyder les atomes d'hydrogène avant leur recombinaison en H2.
Le protocole expérimental se déroule en cinq étapes:
Etape n°1: Le compartiment de détection est mis sous atmosphère d'argon.
Etape n°2: La solution de soude décimolaire (NaOH 0,1 M) est désaérée dans une cellule annexe pendant plusieurs heures.
Etape n°3: La solution de soude est transférée à l'abri de l'air dans la cellule de détection.
Etape n°4: La face de sortie de l'échantillon est traitée suivant un protocole d'oxydation [36].
Etape n°5: La cellule de chargement est remplie avec la solution à étudier.
Le principe et le montage de la cellule peuvent être résumés par le schéma suivant:
Principe de fonctionnement de la cellule de Devanathan :

Montage expérimental:
II ) 2Méthodes électrochimiques
Les méthodes électrochimiques utilisées sont des méthodes transitoires qui consistent à faire varier un paramètre électrique pendant un temps t et à mesurer la réponse d'un système soumis à la perturbation.
II ) 2.1 Voltamétrie cyclique
La voltamétrie cyclique permet de situer qualitativement les différents processus électrochimiques qui peuvent se produire dans une zone de potentiel. La méthode consiste à imposer à l'électrode de travail, un balayage en potentiel en fonction du temps et à mesurer simultanément l'intensité du courant résultant des réactions électrochimiques. La courbe présentant la variation de l'intensité en fonction du potentiel appliqué est appelée voltamogramme. Deux types de voltamogrammes sont susceptibles d'être obtenus selon la nature chimique de l'électrode, la composition de l'électrolyte et le domaine de potentiels exploré: les voltamogrammes dits simples présentant un seul pic dans la partie cathodique et dans la partie anodique (cf. voltamogramme 34a ), et les voltamogrammes dits complexes avec plusieurs pics ou épaulements (cf. voltamogramme 7) La forme des voltamogrammes donne une vue d'ensemble des processus électrochimiques qui se produisent à l'électrode.
Le potentiel de corrosion de l'acier dans le chlorure d'ammonium étant proche de -660 mV/ecs, nous effectuons les balayages en voltamétrie cyclique à partir d'un potentiel de –700 mV/ecs jusqu'à –1700 mV/ecs afin d'éviter la dissolution du fer dans la solution du dépôt.
II ) 2.2 Polarisation potentiostatique
C'est la méthode qui est appelée chronoampèrométrie. Elle consiste à imposer une perturbation en potentiel pendant un temps t et à enregistrer l'évolution du courant pendant le temps t. La valeur du potentiel E1 est choisie (comme en voltamétrie) cathodique par rapport au potentiel de corrosion du substrat et anodique par rapport au potentiel de réduction des espèces électroactives. La valeur du potentiel E2 et la durée t de la polarisation sont fixées en fonction de la réaction cathodique étudiée. Le graphe obtenu est appelé transitoire courant/temps (cf. Figure: Principes des méthodes électrochimiques). La partie (I) de ce transitoire correspond à la charge de la double couche ainsi qu'à la formation des premiers germes. L'augmentation du courant (II) correspond à la croissance des germes formés précédemment (cas de germination instantanée) ou à la formation et la croissance de nouveaux germes (cas de germination progressive). L'analyse de la partie ascendante des transitoires permet de déterminer:
![]() le nombre et la
cinétique de formation des germes (instantanée ou progressive),
le nombre et la
cinétique de formation des germes (instantanée ou progressive),
![]() la nature et la
géométrie de la croissance des germes: monocouche (2D), couche par couche,
tridimensionnelle (3D), conique ou hémisphérique,
la nature et la
géométrie de la croissance des germes: monocouche (2D), couche par couche,
tridimensionnelle (3D), conique ou hémisphérique,
![]() le contrôle cinétique
(transfert de charge ou de masse),
le contrôle cinétique
(transfert de charge ou de masse),
![]() le recouvrement des
centres de croissance.
le recouvrement des
centres de croissance.
La partie (III) correspond à la diminution du courant vers sa valeur stable qui est le courant limite.
II ) 2.3 Polarisation galvanostatique
Cette méthode consiste à imposer une densité de courant constante sur la cathode à revêtir pendant un temps t et à enregistrer l'évolution du potentiel.
Les courbes Potentiel-Temps présentent la variation de la tension d'électrode en fonction du temps pour une certaine intensité du courant imposée. L'analyse de ces courbes permet, pour une réaction simple dont la cinétique est contrôlée par la diffusion des espèces électroactives, de déterminer les paliers de potentiels correspondant aux étapes de la réduction.
Les dépôts massifs sont obtenus en polarisation galvanostatique. Afin d'obtenir un revêtement électrolytique d'épaisseur (e), il est nécessaire d'imposer une densité de courant (i) pendant un temps (t) calculé à partir de la loi de Faraday:

t : Temps (s)
n : Nombre
d'électrons mis en jeu ![]() : Masse
volumique (g cm-3)
: Masse
volumique (g cm-3)
e : Epaisseur du dépôt (cm) S : Aire de la cathode (cm2)
M : Masse molaire (g mol-1) i : Intensité du courant imposé (A)
Les expériences menées en voltamétrie cyclique, polarisations galvanostatique et potentiostatique sont programmées et traitées par le logiciel M270 de EG&G.
II ) 2.4 Impédance électrochimique
En électrochimie, la technique de mesure d'impédances complexes est très utilisée pour l'étude des caractéristiques des diverses interfaces électrode/solution [39]. Le principe général consiste à appliquer à une électrode un stimulus électrique ( tension ou courant) et à mesurer la réponse. L'appareillage utilisé nécessite, qu'en fonction du domaine de fréquences étudiées, deux méthodes de mesure d'impédance du système soient utilisées.
² La première méthode (mode synchrone) consiste à surimposer à un potentiel stationnaire, une tension sinusoïdale de faible amplitude (|dE|) et de fréquence variable. La réponse en intensité donnée par le système est aussi sinusoïdale et d'amplitude |dI| avec un déphasage f par rapport à la tension sinusoïdale tel que tan(f)=-Z'/Z". A chaque fréquence d'excitation correspond une valeur d'impédance qui est définie par son module |Z| = |dE|/|dI| et son déphasage f.
² La seconde méthode (mode transformée de Fourier) consiste à appliquer un bruit blanc (signal v(t)) et à mesurer le courant résultant. On applique ensuite une transformée de Fourier à la mesure pour obtenir une réponse en fréquence et déduire l'impédance. Cette méthode permet une collecte rapide des résultats car un signal unique est appliqué à l'interface pendant un temps très court.
Un diagramme de Nyquist peut être construit en présentant, pour chaque fréquence d'excitation, la partie imaginaire de l'impédance (-Z'') en fonction de sa partie réelle (Z').
Le logiciel M388 de EG&G est utilisé pour piloter et traiter la mesure d'impédance électrochimique à l'aide d'un détecteur synchrone M5208 interfacé à un potentiostat galvanostat PAR273. Ce logiciel permet également de présenter, dans les différents modes, les mesures effectuées.
A partir de ces mesures et en considérant que le comportement électrique d'une cellule électrochimique peut être modélisé par un circuit électrique, nous pouvons caractériser l'interface. En effet, les données expérimentales peuvent être comparées au résultat d'une simulation mathématique basée sur une théorie et un modèle physique (voir organigramme). Si la corrélation est suffisante, on peut déduire les valeurs de chaque composant du circuit équivalent. Généralement le circuit est constitué par des résistances, des capacités, des éléments à phase constante (CPE) et des éléments inductifs. L'élément à phase constante est défini par :
![]()
est représentatif d'une série d'éléments en fonction des valeurs de n. Pour n=-1 la CPE représente une inductance, pour n=0 une résistance pure, pour n=1/2 une impédance de Warburg, et pour n=1 une capacité. L'élément à phase constante s'utilise également pour décrire un comportement diélectrique non idéal. La partie modélisation est effectuée à l'aide du logiciel EQUIVCRT de Boukamp [40]. Ce logiciel permet, par une méthode d'ajustement de paramètres, de déterminer les grandeurs du circuit équivalent utilisé pour modéliser le comportement électrique de la cellule électrochimique.
Organigramme de
caractérisation d'un système substrat/électrode. [41]
Durant ce travail, la technique de mesure d'impédance électrochimique a été utilisée de deux manières différentes:
![]() L'utilisation principale
consiste à la caractérisation de revêtement,
L'utilisation principale
consiste à la caractérisation de revêtement,
![]() une autre utilisation
permet de suivre l'évolution d'une capacité en fonction du temps.
une autre utilisation
permet de suivre l'évolution d'une capacité en fonction du temps.
Caractérisation des revêtements
Le dispositif expérimental utilisé est constitué par une cellule classique à trois électrodes. Les revêtements sont étudiés en solution de chlorure de sodium 0,5M. La durée d'immersion avant le début de l'expérience est fixée à une heure afin d'obtenir un quasi état stationnaire. L'amplitude de la perturbation est de 2 mV autour du potentiel de corrosion. Le diagramme d'impédance est obtenu en utilisant les deux modes de mesure. Pour les fréquences comprises entre 10 kHz et 10 Hz, les mesures se font en mode synchrone. Pour le domaine de fréquences 10 Hz à 10 mHz, on utilise le mode transformée de Fourier.
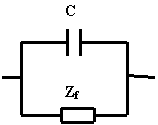
Evolution de la capacité interfaciale en fonction du temps
Une utilisation spécifique de la mesure d'impédance est mise en œuvre pour déterminer l'évolution de la capacité interfaciale du système en fonction du temps. Etant donné la durée d'une mesure d'impédance sur tout le domaine de fréquence, il n'est pas possible d'utiliser le protocole décrit précédemment pour suivre l'évolution d'un système rapide. Il faut donc faire une approximation pour déterminer rapidement la valeur de la capacité de double couche. Nous considérons que l'interface électrochimique du système peut être modélisée par la représentation suivante:
C :
Capacité de double couche Zf:
Impédance faradique

L'expression de l'impédance du système peut être calculée:
![]()
![]() C'est à dire :
C'est à dire :
Quand la pulsation w est faible, Z tend vers Zf, le circuit est essentiellement faradique.
Quand la pulsation w est grande, Z tend vers 1/jC w, le circuit est essentiellement capacitif.
Une mesure à haute fréquence (1 kHz), permet donc d’avoir une mesure rapide de la capacité. Le suivi de l'évolution de la capacité de double couche en fonction du temps peut ainsi être effectué par une succession, à intervalle régulier, de mesures hautes fréquences.

II ) 3Méthodes non électrochimiques
Diverses méthodes ont été utilisées, l'absorption atomique: la fluorescence X, la microscopie électronique à balayage et la microanalyse X qui ne seront pas décrites ici car elles sont bien connues.
II ) 3.1 Diffraction de rayons X
La diffraction de rayons X permet de déterminer les phases présentes dans les dépôts. Cette technique est utilisée selon le montage conventionnel (q - 2q) et en incidence rasante. L'appareil de mesure est le D 5000 de SIEMENS.
Les raies caractéristiques utilisées sur le D 5000, proviennent des transitions électroniques d'une anticathode de cuivre:
lk-L2 = 1,5405 Å lk-L3 = 1,5443 Å lk-M = 1,3922 Å
La raie k-L2 est accompagnée des raies k-L3 et k-M, d'intensité notable. On obtient des raies supplémentaires de diffraction correspondant aux différentes longueurs d'onde des rayons X. La loi de Bragg qui rend compte de cette réflexion, indique que chaque radiation contenue dans le diagramme de diffraction engendre son propre diagramme. Il est donc impératif de monochromatiser au mieux le faisceau.
nl = 2 dhkl sin q avec l : longueur d'onde des rayons X
n : ordre de réflexion
dhkl : distance réticulaire de la famille des plans (hkl).
Le faisceau peut être monochromatisé par deux méthodes, l'une approchée : filtre b, l'autre rigoureuse: monochromateur. Le diffractomètre D 5000 est équipé d'un monochromateur plan arrière monté en configuration parallèle. Il est placé entre l'échantillon et le détecteur. Lorsque l'échantillon contient un élément lourd, il faut éviter d'utiliser comme anticathode un élément de numéro atomique juste supérieur, pour s'affranchir d'un phénomène de fluorescence. L'emploi d'un diffractomètre muni d'un monochromateur arrière évite d'avoir à changer l'anticathode. Le détecteur installé sur le D5000 est un détecteur solide à scintillation qui permet de discriminer les énergies. Les diagrammes de diffraction de rayons X comportent le nombre de coups détectés en fonction de l'angle (q - 2q). La position de l'intensité de chaque raie permet de caractériser l'échantillon ou de déterminer les orientations préférentielles des plans de symétries du réseau cristallin. L'utilisation d'un logiciel approprié permet de calculer les paramètres de la maille élémentaire.
La détermination des phases cristallines se fait habituellement par comparaison des diagrammes de diffraction obtenus avec la base de données JCPDS (Joint Comittee on Powder Diffraction Standard). Les méthodes d'analyses par rayons X font intervenir une profondeur de pénétration des rayons X comprise entre le micromètre et le millimètre. Par contre, en incidence rasante, cette profondeur irradiée diminue proportionnellement avec l'angle d'incidence a du faisceau de rayons X. Ainsi les méthodes d'analyses par rayons X permettent l'étude des couches minces jusqu'à quelques couches atomiques. La diffraction de rayons X en configuration q-2q est utilisée pour caractériser les dépôts épais et la diffraction de rayons X en mode rasant pour caractériser la surface des échantillons après corrosion.
II ) 3.2 Microscopie à effet tunnel et microscopie à force atomique
La microscopie à effet tunnel (Scanning Tunneling Microscopy) et la microscopie à force atomique (Atomic Force Microscopy) sont deux techniques qui permettent d'obtenir une image de la surface des matériaux avec une résolution pouvant atteindre l'échelle atomique. Basées sur deux phénomènes physiques différents, ces deux techniques utilisent le même principe général de fonctionnement qui est résumé par le schéma suivant:
La
microscopie à effet tunnel
La microscopie à effet tunnel est basée sur un phénomène physique connu depuis les origines de la mécanique quantique. Cette technique inventée en 1981 a valu à ses inventeurs G. Binning et H Roher le Prix Nobel de Physique en 1986 [42]. La microscopie à effet tunnel exploite le passage d'électron par effet tunnel entre une pointe conductrice très fine et un échantillon soumis à une différence de potentiel. Le courant provenant de l'effet tunnel varie exponentiellement avec la distance entre la sonde et l'échantillon. Ce type de relation permet de donner une image de la surface avec une très grande précision. La résolution verticale peut être inférieure à l'angström, la résolution latérale est à l'échelle atomique. Le balayage de la surface peut s'effectuer selon deux modes. En mode hauteur constante, la pointe balaie la surface dans un plan horizontal et on mesure la variation de l'intensité du courant dû à l'effet tunnel qui est fonction de la topographie et des propriétés électroniques de la surface. En mode courant constant, le microscope ajuste la hauteur du scanner pour garder l'intensité du courant d'effet tunnel constante. Dans ce cas c'est le mouvement du scanner qui donne une image de la topographie de l'échantillon. La microscopie à effet tunnel n'est possible qu'avec des échantillons conducteurs contrairement à la microscopie à force atomique qui peut s'effectuer sur des échantillons non conducteurs.
Microscopie
à Force Atomique
La méthode est basée sur le déplacement d'une micropointe liée à un bras flexible à la surface d'un échantillon. Les forces de Van der Waals générées par l'interaction entre la micropointe et les atomes de la surface provoquent la déformation de ce bras. Un détecteur mesure les mouvements du bras et génère une image de la topographie de la surface sur l'ordinateur.
Le microscope Autoprobe CP de Park Scientific Instrument peut fonctionner en utilisant ces deux modes de fonctionnement. Proscan est le logiciel de traitement d'image utilisé.
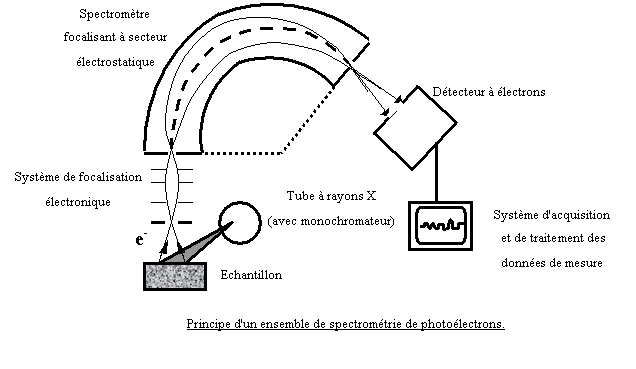
II ) 3.3
Spectrométrie de photo électron de
rayons X (XPS)[43]
Les premiers travaux sur la spectrométrie des photoélectrons ont été publiés en Angleterre vers 1914 par Robinson et en France, en 1921, par M. de Broglie. La spectrométrie des électrons est une méthode d'analyse des surfaces. Son principe est fondé sur l'interaction d'un rayonnement primaire fourni par un tube à rayon X avec un matériau qui libère des électrons issus des niveaux atomiques excités. A la sortie du matériaux irradié, certains de ces électrons ont une énergie caractéristique de leur niveau d'origine. Par leur spectrométrie, on peut alors les utiliser pour identifier les atomes, ainsi que pour mesurer les niveaux d'énergie de ces derniers. Les électrons émis ont généralement une énergie faible et sont donc fortement absorbés. Ceux qui arrivent à la surface, avec une énergie suffisante pour fournir le travail de sortie, ne proviennent que d'une mince couche superficielle. Connaissant les niveaux d'énergie caractéristiques de tous les éléments par des tables, on peut les identifier par leur spectre de photoélectrons. Tous les éléments, y compris les éléments légers à partir du lithium, peuvent être analysés.
Les ordres de grandeurs limites de détection sont:
Concentration limite détectable 10-3
Masse minimum détectable 10-11 g
Le spectre de photoélectrons comprend des pics provenant des niveaux atomiques extérieurs dont l'énergie est sensible à l'état de liaison chimique des atomes et à leur environnement. La position du pic de photoélectron, d'un niveau donné d'un élément, dépend donc directement de son état chimique. En se rapportant à la position des pics donnés par des éléments purs pris comme témoins, la mesure du déplacement d'un pic, dit déplacement chimique, apporte des informations sur l'état de liaison à la surface d'un solide: par exemple, le degré d'oxydation de métaux, la coordinence, etc. .
La figure suivante représente le schéma de principe courant d'un appareillage XPS.
Les électrons passant par le diaphragme de sortie du spectromètre produisent un signal électrique du détecteur qui est amplifié, puis enregistré en fonction du champ dispersif, pour fournir le spectre I(E) des électrons émis par l'échantillon. Toute la partie de l'appareil parcourue par les électrons est sous un vide poussé (10-6 à 10-9 torr).
II ) 3.4 Caractérisation de la composition chimique et de l'épaisseur des revêtements
Pour caractériser les échantillons, il faut déterminer les méthodes les plus adéquates. Dans un premier temps, nous voulons caractériser deux propriétés liées aux dépôts :
![]() la teneur en Manganèse
la teneur en Manganèse
![]() l'épaisseur du dépôt.
l'épaisseur du dépôt.
Pour déterminer ces propriétés, différentes techniques sont disponibles et il faut sélectionner celles qui répondent le mieux à la détermination de ces paramètres.
![]() Pour doser la teneur en
manganèse, trois techniques peuvent être utilisées. L'absorption atomique qui
permet de réaliser un dosage très précis des éléments contenus dans le dépôt.
C'est une méthode quantitative plus précise que la microanalyse X, mais elle
est en revanche destructive. La microanalyse X qui est une méthode semiquantitative
et enfin la fluorescence X.
Pour doser la teneur en
manganèse, trois techniques peuvent être utilisées. L'absorption atomique qui
permet de réaliser un dosage très précis des éléments contenus dans le dépôt.
C'est une méthode quantitative plus précise que la microanalyse X, mais elle
est en revanche destructive. La microanalyse X qui est une méthode semiquantitative
et enfin la fluorescence X.
![]() Pour évaluer l'épaisseur
du dépôt, la méthode la plus simple est la double pesée. Une autre méthode
consiste à faire une coupe transversale du dépôt puis un enrobage, un polissage
et enfin une observation micrographique de la tranche. La dernière méthode
consiste à faire une mesure d'épaisseur par fluorescence X.
Pour évaluer l'épaisseur
du dépôt, la méthode la plus simple est la double pesée. Une autre méthode
consiste à faire une coupe transversale du dépôt puis un enrobage, un polissage
et enfin une observation micrographique de la tranche. La dernière méthode
consiste à faire une mesure d'épaisseur par fluorescence X.
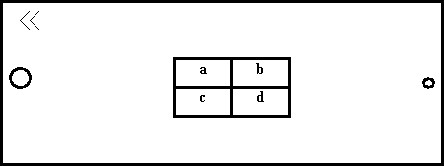
Pour tester ces différentes techniques, nous utilisons une série d'échantillons pour éprouver ces différentes techniques. Quatre échantillons sont découpés dans la même plaque de la manière suivante.

Deux sont destinés à l'absorption atomique (a et b), un à un examen en coupe micrographique (c), le dernier (d) aux méthodes non destructrices (microanalyse et Fluorescence X).
Comparaison des résultats avec les différentes méthodes
Le tableau ci-dessous groupe les résultats.
|
Echantillon |
j mAcm-2 |
e mm Théorique |
Masse du dépôt g |
e mm Calculée |
e mm Coupe |
e mm Fluo X |
% Mn A. A* |
% Mn MicroX* |
|
1 |
30 |
1 |
0,0030 |
0,5 |
** |
0,4 |
5,60 |
4,35 |
|
1' |
30 |
1 |
0,0031 |
0,5 |
|
0,4 |
5,77 |
4,35 |
|
2 |
20 |
10 |
0,0301 |
5,4 |
5,5 |
5,4 |
3,32 |
2,97 |
|
2' |
20 |
10 |
0,0298 |
5,2 |
|
5,4 |
3,36 |
2,97 |
|
3 |
20 |
1 |
0,0035 |
0,5 |
** |
0,5 |
3,43 |
2,79 |
|
3' |
20 |
1 |
0,0034 |
0,6 |
|
0,5 |
3,35 |
2,79 |
|
4 |
30 |
10 |
0,0322 |
5,6 |
5,5 |
5,5 |
7,92 |
8,44 |
|
4' |
30 |
10 |
0,0310 |
5,4 |
|
5,5 |
7,90 |
8,44 |
* Les pourcentages de manganèse sont exprimés en masse.
** Indéterminée
Comparaison
des résultats de différentes méthodes de caractérisation.
Mesure
de la teneur en Manganèse
L'écart entre les mesures effectuées par microanalyse X et absorption atomique est de +/- 1 % de manganèse. Les mesures obtenues en fluorescence X sont par contre aberrantes (non reportées dans le tableau) car la résolution en fluorescence X est trop faible pour pouvoir séparer le manganèse du fer (substrat) comme l'indique le tableau suivant.
|
Symbole atomique |
Mn |
Fe |
Zn |
|
N° atomique |
25 |
26 |
30 |
|
Masse atomique |
54,94 |
55,85 |
65,38 |
|
densité gcm-3 |
7,20 |
7,87 |
7,13 |
|
Situation du pic en énergie KeV |
5,90 |
6,40 |
8,64 |
|
N° du canal |
45 |
49 |
66 |
|
Début du spectre |
36 |
40 |
53 |
|
Fin du spectre |
54 |
56 |
82 |
Constantes physiques des spectres en fluorescence X.
En effet les pics sont trop proches l'un de l'autre pour être efficacement distingués. Les pics du zinc par contre, se trouvent dans une autre fenêtre d'énergie. Généralement, le faible pouvoir de résolution peut être compensé par un traitement mathématique du spectre. Cependant le logiciel de déconvolution de pic n'est pas paramétré pour traiter le pic du manganèse. Nous obtenons donc des pourcentages de manganèse qui sont directement fonction de l'épaisseur du revêtement et non pas de la valeur intrinsèque de la teneur en manganèse. La mesure s'avère également fausse avec des dépôts plus épais.
Nous choisissons donc de déterminer la teneur en manganèse par microanalyse X. C'est une méthode non destructive qui donne un ordre de grandeur à 1 ou 2 % du taux de manganèse.
Mesure
d'épaisseur
En comparant les résultats de mesure d'épaisseur issus des trois techniques (coupe transversale, différence de masse, fluorescence X), nous trouvons des résultats proches à 0,5 mm près. La méthode de fluorescence X est simple à mettre en œuvre et permet de mesurer ponctuellement l'épaisseur de tout le dépôt et d'en dresser un profil. Les autres méthodes donnent une information strictement ponctuelle (coupe transversale) ou moyenne (différence de masse). C'est donc la fluorescence X qui est utilisée pour déterminer l'épaisseur des dépôts et la microanalyse X pour déterminer leur teneur en manganèse.
II ) 4Conditions d'élaboration et caractérisation des revêtements
Les conditions d'élaboration des revêtements étudiés sont présentés ultérieurement sous forme de tableau. Les caractéristiques des échantillons caractérisés après élaboration sont groupées dans le tableau 1 Les caractéristiques des revêtements caractérisés après exposition en corrosion naturelle sont données dans le tableau 2 et tableau 3.