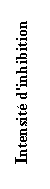|
L |
a mise au point d’un revêtement anticorrosion
présente un intérêt économique et technique. Les coûts et les conséquences
engendrés par la corrosion (perte de métaux, protection anticorrosion, maintenance)
sont estimés à 3% du produit national brut dans les pays industrialisés. Parmi
ces conséquences, on peut citer, par exemple, la destruction prématurée des
structures en acier soumises à l’action corrosive de l’eau et de l’air en
atmosphères chlorées ou soufrées. Depuis plus de soixante ans, la protection
des aciers contre la corrosion est assurée par le dépôt d’un métal à faible
coût, le zinc qui lui confère une bonne résistance à la corrosion. De
nombreuses branches d’activités industrielles (le bâtiment, le génie civil,
l’électroménager, l’automobile etc.) utilisent l’acier galvanisé car après
revêtement, la pièce peut être forgée, soudée, phosphatée et peinte. L’activité
protection contre la corrosion, à elle seule, consomme environ 35% de la production
de zinc.
L’élaboration d’un dépôt de zinc peut se faire
selon deux types de procédés : au trempé dans un bain de zinc fondu ou par
électrocristallisation. Le choix est déterminé en fonction des caractéristiques
que l'on veut donner aux dépôts et des épaisseurs désirées. Les tôles utilisées
dans le secteur automobile nécessitent des mises en formes (emboutissages,
pliages) qui rendent indispensables un contrôle rigoureux de la structure et de
l’épaisseur du dépôt. Les dépôts ont une épaisseur de l’ordre de 10 à 30 mm selon la fonction ou
la position de la pièce sur le véhicule. De tels dépôts ne peuvent pas
s’obtenir par galvanisation à chaud car l’épaisseur minimale accessible est de
50 mm. Les
dépôts sont donc élaborés par électrocristallisation. Cette technique permet un
parfait contrôle de l’épaisseur pour ces ordres de grandeur.
Le dépôt de zinc
joue, dans un premier temps, un rôle un protection physique. En effet, en
fonction du milieu d’exposition, il forme différents types de produits de
corrosion qui possèdent des propriétés intéressantes. En atmosphère sèche et à
température ambiante, une très mince couche d’oxyde se forme rapidement. Dans
l’air humide, c'est une couche de produits insolubles composés de carbonate de
zinc (ZnCO3) (dont la forme naturelle est la smithonite),
d’hexahydroxydicarbonate de pentazinc et de l’hydroxyde de zinc Zn(OH)2
sous diverses formes allotropiques. Dans l’eau de mer, apparaissent des sels de
zinc tels que la simonkolléite ZnCl24(ZnOH)2. Dans les
trois milieux, les produits de corrosion forment une couche protectrice qui
ralentit la dissolution du zinc. Ainsi, l’acier est isolé du milieu agressif
par une couche de zinc et une couche de produits de corrosion. Autre point
important, en cas de mise à nu du substrat ferreux, le zinc peut, contrairement
à un revêtement organique, colmater la blessure par la formation des ces
oxydes. Lorsqu'une blessure du dépôt de zinc met l'acier à nu, le zinc joue un
autre rôle. Pour le comprendre, il faut examiner les potentiels
électrochimiques respectifs de chaque métal. Les données numériques sont
données dans le tableau suivant:
|
Couple électrochimique |
Potentiel en mV |
|
Electrode saturée au calomel (KCl) |
241 |
|
H+/H2 |
0 |
|
Ni2+/Ni |
-230 |
|
Co2+/Co |
-280 |
|
Fe2+/Fe |
-409 |
|
Zn2+/Zn |
-762 |
|
Mn2+/Mn |
-1029 |
Potentiels électrochimiques théoriques.
Le zinc présente
un potentiel électrochimique théorique inférieur de 353 mV par rapport au fer. En
cas de contact entre les deux métaux, le zinc s'oxyde (anode sacrificielle) et
le fer (cathode) se trouve protégé.
e- e- e-
 Nous pouvons résumer les trois effets
protecteurs du zinc par les schémas suivants:
Nous pouvons résumer les trois effets
protecteurs du zinc par les schémas suivants:

Protection physique Colmatage de blessure Protection cathodique
Différents
aspects de la protection du zinc sur un substrat ferreux.
Depuis les années soixante, la demande de
protection anticorrosion des carrosseries automobiles augmente fortement. Les
dépôts de zinc ne suffisant plus à assurer une garantie suffisante contre la
corrosion pour des durées de plus de trois ans, l’ensemble des constructeurs
ont axé leurs recherches sur des revêtements électrodéposés de plus en plus
performants permettant de plus en plus d’applications. Une voie consiste à
développer les systèmes alliés à base de zinc qui présentent une résistance à
la corrosion supérieure aux dépôts de zinc. Ainsi le zinc-nickel, le zinc-fer,
et le zinc-cobalt reçoivent un vif intérêt dans le milieu industriel car ils
permettent de très bonnes finitions (phosphatations, chromatations,
cosmétiques…) et des applications spécifiques (adhérisation de caoutchouc…).
Pour comprendre l'intérêt de l'utilisation des
zincs alliés, il faut considérer deux aspects. Le premier aspect concerne la
modification de la cinétique d'électrocristallisation par l'introduction d'un
second métal. Cet aspect est difficile à appréhender et nous ne pouvons pas
attribuer à priori de préférence à un métal en particulier. Le deuxième aspect
concerne le rôle du second métal dans le dépôt.
De la même manière que l'on considère la
valeur relative du potentiel du zinc par rapport au fer, il est nécessaire de
considérer la valeur relative du potentiel de l'élément d'alliage par rapport
au zinc. Si on examine les potentiels relatifs des différents couples, nous
constatons que le nickel, le fer, le cobalt ont des potentiels cathodiques par
rapport au zinc. Ces éléments ne peuvent donc pas jouer de rôle sacrificiel par
rapport au zinc. De plus, ils sont susceptibles d'accélérer le processus de
corrosion en induisant des couplages galvaniques qui conduisent à une
dézincification.
Partant de ce constat, une thèse à été initiée
au Laboratoire de Physico Chimie des Matériaux de Marseille par M. Eyraud [1]
concernant l'élaboration d'un dépôt de zinc-manganèse. En effet, le manganèse
présente par rapport au zinc et au fer un intérêt réel sur le plan
électrochimique. Son potentiel électrochimique est en effet inférieur à celui
du zinc. Il est donc susceptible de fournir une protection cathodique à la fois
à l'acier et au zinc. En considérant les lois thermodynamiques, le manganèse
puis le zinc et enfin l'acier doivent être successivement oxydés. Etant donné
la différence entre les potentiels électrochimiques du zinc et du manganèse,
ils ne peuvent pas se déposer simultanément et un additif est nécessaire pour
rapprocher les potentiels d'électrocristallisation. Le citrate en milieu
sulfate a été utilisé dans toutes les études publiées sur le codépôt du zinc et
du manganèse mais M. Eyraud [1] a
montré que ce bain nécessite un contrôle très rigoureux du pH dans un
intervalle de 0,4 unités. L'utilisation de ce type de bain en milieu industriel
semble donc compromise malgré de très bonnes performances du revêtement tant au
point de vue de la tenue à la corrosion que du point de vue de sa soudabilité,
de sa phosphatabilité et de son comportement à la peinture.
La bonne résistance à la corrosion des dépôts
de zinc-manganèse, nous a incité à rechercher un nouveau bain qui permette d'élaborer
industriellement des dépôts Zn-Mn. La présence d'ammonium étant indispensable à
la cristallisation du manganèse, le bain choisi est à base de chlorure
d'ammonium.
![]() La première partie de
cette étude présente rapidement, les caractéristiques générales des métaux
utilisés et l’électrocristallisation.
La première partie de
cette étude présente rapidement, les caractéristiques générales des métaux
utilisés et l’électrocristallisation.
![]() La deuxième partie est
consacrée à la description des dispositifs et des techniques électrochimiques,
ainsi qu’aux méthodes d’analyse.
La deuxième partie est
consacrée à la description des dispositifs et des techniques électrochimiques,
ainsi qu’aux méthodes d’analyse.
![]() La troisième partie est
une étude du rôle des additifs sur la cinétique d’électrocristallisation du
zinc, du manganèse et de l’alliage zinc-manganèse. La présence de certaines
substances organiques, désignées sous le nom général d’additifs, est
indispensable pour obtenir des dépôts de bonne qualité, même dans le cas de
l’électrocristallisation d’un seul métal. Dans le cas de deux métaux à déposer,
la présence de ces additifs permet aussi, en modifiant les surtensions de
cristallisation, d’obtenir une déposition simultanée et donc un alliage. Une
étude cinétique préliminaire tente de comprendre l’action des additifs sur un
mécanisme de réduction plus simple que celle d’un métal, c’est la réduction du
proton. L’intérêt est double car, étant donné les potentiels très actifs du
zinc et du manganèse, la réduction du proton accompagnera toujours leur
électrocristallisation. L'étude de la réduction du proton est par conséquent
nécessaire.
La troisième partie est
une étude du rôle des additifs sur la cinétique d’électrocristallisation du
zinc, du manganèse et de l’alliage zinc-manganèse. La présence de certaines
substances organiques, désignées sous le nom général d’additifs, est
indispensable pour obtenir des dépôts de bonne qualité, même dans le cas de
l’électrocristallisation d’un seul métal. Dans le cas de deux métaux à déposer,
la présence de ces additifs permet aussi, en modifiant les surtensions de
cristallisation, d’obtenir une déposition simultanée et donc un alliage. Une
étude cinétique préliminaire tente de comprendre l’action des additifs sur un
mécanisme de réduction plus simple que celle d’un métal, c’est la réduction du
proton. L’intérêt est double car, étant donné les potentiels très actifs du
zinc et du manganèse, la réduction du proton accompagnera toujours leur
électrocristallisation. L'étude de la réduction du proton est par conséquent
nécessaire.
![]() L’élaboration des dépôts
massifs et leur caractérisation constituent la quatrième partie. Une étude en
cellule de Hull permet de déterminer les paramètres d'élaboration des dépôts.
Leur caractérisation est faite par différents types de microscopie, par
diffraction de rayons X, par spectrométrie XPS. Le comportement à la corrosion
est une des difficultés majeures de la caractérisation. En effet les méthodes
utilisées habituellement sont très difficilement applicables aux dépôts
métalliques et il n’existe pas de méthodes spécifiques. Ainsi, les courbes de
polarisation et la spectrométrie d’impédance électrochimique seront utilisées
pour cette caractérisation.
L’élaboration des dépôts
massifs et leur caractérisation constituent la quatrième partie. Une étude en
cellule de Hull permet de déterminer les paramètres d'élaboration des dépôts.
Leur caractérisation est faite par différents types de microscopie, par
diffraction de rayons X, par spectrométrie XPS. Le comportement à la corrosion
est une des difficultés majeures de la caractérisation. En effet les méthodes
utilisées habituellement sont très difficilement applicables aux dépôts
métalliques et il n’existe pas de méthodes spécifiques. Ainsi, les courbes de
polarisation et la spectrométrie d’impédance électrochimique seront utilisées
pour cette caractérisation.
![]() La dernière partie est
consacrée à l'étude de revêtements exposés en corrosion naturelle.
La dernière partie est
consacrée à l'étude de revêtements exposés en corrosion naturelle.
I )
Généralités
I ) 1 Le zinc
Le tableau
ci-dessous présente les données numériques des propriétés
physico-chimiques du zinc :
|
Symbole |
Zn |
|
Numéro atomique |
30 |
|
Densité |
7,14 |
|
Masse atomique |
65,38 |
|
Rayon atomique |
0,74 Å |
|
Système cristallin |
Hexagonal compact (a = 2,665Å, c = 4,947Å, c/a = 1,8563) |
|
Température de fusion |
419,5°C |
L’électrocristallisation de zinc peut se faire
à partir de plusieurs types de bains de zingage (acide (KCl) , alcalin (NaOH)),
en fonction des caractéristiques de la pièce à traiter (dimensions, formes,
etc.) , de l’épaisseur du dépôt de zinc et de l’aspect que l’on désire, mat ou
brillant. Dans tous les cas on utilise des additifs qui permettent d'obtenir
des dépôts de bonnes qualités [2-11].
I ) 2 Le manganèse
Très voisin du fer par ses propriétés
physico-chimiques, le manganèse est extrêmement répandu. La connaissance de la
magnésie noire (bioxyde de manganèse naturel) remonte à la plus haute
antiquité: Pline mentionne son emploi dans la fabrication du verre. Le
manganèse peut être obtenu par réduction des oxydes ou par électrolyse. Le
manganèse, à l'état pur, ne présente pas de propriétés physiques ou mécaniques
intéressantes et de ce fait n'est pas utilisé à l'état pur. Par contre, il
constitue un élément d'addition appréciable dans les alliages (acier et non
ferreux).
I ) 2.1 Propriétés physico-chimiques du manganèse
Le tableau
ci-dessous présente les données numériques des propriétés
physico-chimiques du manganèse :
|
Symbole |
Manganèse |
|
Numéro atomique |
25 |
|
Masse atomique |
54,938 |
|
Température de fusion |
1244°C |
Le manganèse cristallise d'une manière
complexe sous la forme de quatre variétés allotropiques [12-14]. Chaque phase
offre des caractéristiques spécifiques et un domaine de température d'existence
propre. Ces diverses phases du manganèse sont : alpha (stable jusqu'à 700°C),
bêta (entre 700 et 1079°C), gamma (entre 1079 et 1143°C) et delta (entre 1143°C
et la température de fusion).
Variété allotropique a
Stable à la température ambiante, elle a une
masse volumique de 7,44 gcm-3. Sa structure dérive du système
cubique centré avec un paramètre a = 8,944 Å. A chaque point de son réseau est
associé un groupe de 29 atomes. La maille élémentaire comprend ainsi 58 atomes.
Autour de chaque atome du réseau cubique
centré sont disposés :
![]() 4 atomes A formant un tétraèdre,
4 atomes A formant un tétraèdre,
![]() 12 atomes D1 formant un
polyèdre à 14 faces cubiques et octaédriques,
12 atomes D1 formant un
polyèdre à 14 faces cubiques et octaédriques,
![]() 12 atomes D2 formant un
octaèdre à 4 faces triangulaires et 4 faces hexagonales.
12 atomes D2 formant un
octaèdre à 4 faces triangulaires et 4 faces hexagonales.

Etant donné sa structure complexe la variété a du manganèse présente
peu de plans de glissement et se caractérise par sa fragilité. La structure du
manganèse a est schématisée par la figure ci-dessous:
 C'est un réseau cubique
complexe comportant 20 atomes par maille élémentaire dont le paramètre (a) vaut
6,289 Å. Les 20 atomes sont disposés en deux groupes, l'un de 12 atomes (type
I), l'autre de 8 atomes (type II). Sa masse volumique vaut 7,299 gcm-3.
La structure du manganèse b est schématisée par la figure ci-dessous:
C'est un réseau cubique
complexe comportant 20 atomes par maille élémentaire dont le paramètre (a) vaut
6,289 Å. Les 20 atomes sont disposés en deux groupes, l'un de 12 atomes (type
I), l'autre de 8 atomes (type II). Sa masse volumique vaut 7,299 gcm-3.
La structure du manganèse b est schématisée par la figure ci-dessous:

Elle présente deux structures en fonction de la
température. A température élevée, la structure est cubique à faces centrées
simple (a = 3,86 Å). A température ambiante, la structure est quadratique à
faces centrées simple (a = 3,776 Å) et le rapport des paramètres c/a = 0,937).
Ces structures ainsi que leurs domaines de stabilité ont été déterminés par
extrapolation. La masse volumique est de 7,21 gcm-3.
Appartenant au système cubique centré simple
(a= 3,075 Å à 1140°C et 3,057 Å à 1244°C), la forme d est une variété de haute température.
Transformation
allotropiques du manganèse
Entre les différentes variétés allotropiques
du manganèse, peuvent avoir lieu les transformations suivantes :
700°C 1079°C 1143°C 1244°C
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]() a b g d Liquide
a b g d Liquide
 métastable
métastable
dés
la température ambiante
Le tableau suivant reprend les données
relatives aux propriétés des quatre variétés allotropiques du manganèse :
|
Formes allotropique du Manganèse |
a |
b |
g |
d |
|
Structure cristalline |
c.c. Complexe |
c. Complexe |
c.f.c./t.f.c. |
c.c. |
|
Nombre d'atomes par maille |
58 |
20 |
4 |
2 |
|
Paramètre cristallin (Å) |
8,944 |
6,289 |
3,86 a=3,766 c/a=0,937 |
3,066 |
|
Température de transformation (°C) |
700 |
1079 |
1143 |
1244 |
|
Densité à 20°C (gcm-3) |
7,43 |
7,29 |
7,18 |
6,3 |
|
Potentiel Standard (mV/ENH) |
-1058 |
* |
-1085 |
* |
Ce tableau indique deux potentiels standards du couple Mn/Mn2+.
En effet par électrolyse en solution aqueuse, on obtient soit le manganèse
alpha dur et fragile, soit le manganèse gamma, ductile. De plus, certains
travaux précisent qu'il se forme au début de l'électrolyse une couche amorphe de
manganèse a qui présente une faible surtension d'hydrogène. Au cours de
l'électrolyse, le dégagement intense d'hydrogène provoque une alcalinisation de
la couche de l'électrolyte voisine de la cathode. Le potentiel d'électrode
décroît ainsi jusqu'à une valeur inférieure au potentiel d'équilibre du
manganèse g, où le manganèse se dépose sous cette forme allotropique. Le manganèse
gamma se transforme d'une manière irréversible en manganèse alpha au cours du
temps, à une vitesse qui dépend de la température.
I ) 2.2 Electrocristallisation du manganèse
Le manganèse est le métal le moins noble
obtenu par électrolyse en solutions aqueuses. Son élaboration est délicate et
exige un certain nombre de précautions. L'électrolyte de base, constitué à
partir de sel de manganèse, ne doit pas contenir d'impuretés métalliques.
Celles ci se déposent préférentiellement au manganèse et sont susceptibles soit
de provoquer une pollution du dépôt avec formation de couple galvanique, soit
d'empêcher tout dépôt (cas du cobalt, de l'argent, du cuivre, du nickel).
Durant les années quarante, Dean [15] puis l' "US bureau of Mines"
ont développé un procédé d'extraction du manganèse par électrolyse qui a obtenu
un grand succès. Les solutions chlorhydriques ou sulfuriques sont les plus
utilisées, elles se caractérisent dans tous les cas par la présence d'ammonium
dont le rôle est déterminant.
Rôle de l'ammonium [14 et ses références]
Le cation ammonium intervient en concentration
minimale de 20 g l-1 pour trois raisons :
![]() l'effet tampon du couple
NH4+/NH3
l'effet tampon du couple
NH4+/NH3
![]() l'activation de la
cathode
l'activation de la
cathode
![]() la conductivité du bain.
la conductivité du bain.
Détaillons chacun de ces aspects.
![]() Effet tampon du couple NH4+/NH3
Effet tampon du couple NH4+/NH3
Le couple NH4+/NH3 confère
au bain une propriété tampon dans le domaine de pH légèrement basique (7-8) qui
est habituellement celle des catholytes d'extraction du manganèse. Un pH plus
faible favorise la réduction du proton au détriment de la réduction du cation
manganeux occasionnant ainsi une diminution du rendement de courant du dépôt de
manganèse. Un pH plus élevé favorise la précipitation de l'hydroxyde de
manganèse qui risque d'être inclus dans le dépôt. L'accroissement de la
concentration en cations ammonium et/ou manganeux dans l'électrolyte améliore
son pouvoir tampon. La concentration de l'ammoniac est liée au pH par
l'équilibre :
NH4+ ó NH3 +
H+ (1)
Soit encore:
NH4+ + OH- ó NH3 +
H2O (2)
Les cations
manganeux modifient la courbe de neutralisation d'une solution d'ammonium. En
effet, en solution aqueuse, il existe entre NH3 et Mn2+
une interaction qui conduit à la formation de complexes aminés suivant la
réaction:
[Mn(H2O)x]2+
+ n NH3 ó [Mn(NH3)n(H2O)x-n]2+
+ nH2O (3)
Cette réaction déplace
vers la droite l'équilibre des réactions (1) et (2) de formation de NH3
et fait décroître le pH de la solution. L'électrolyte peut être ainsi considéré
comme un système Mn2+/NH4+/NH3/H2O
dans lequel les complexes [Mn(NH3)n(H2O)x-n]++
empêchent la précipitation d'hydroxyde de manganèse suivant la réaction :
Mn2+ + 2OH- ó Mn
(OH)2 (4)
Pour une solution
dont le pH est inférieur à 10, le Mn(OH)2 précipite si la
concentration d'ammonium est insuffisante, ce qui peut s'expliquer par les réactions
suivantes:
![]() Dissociation de
l'ammoniaque si le rapport [NH4+ ] /[ Mn2+ ]
est inférieur à 1.
Dissociation de
l'ammoniaque si le rapport [NH4+ ] /[ Mn2+ ]
est inférieur à 1.
Mn2+ +
n(NH4OH) + b NH4+ ó Mn(OH)2 +(n-2) NH4OH
+ (b+2) NH4+ (5)
![]() Complexation du cation
Mn2+ si ce rapport est
supérieur à 1.
Complexation du cation
Mn2+ si ce rapport est
supérieur à 1.
Mn2+ + n(NH4OH) + a NH4+ ó Mn(NH3)n++ + nH2O + a NH4+ (6)
Pendant
l'électrolyse du manganèse, le dégagement d'hydrogène provoque une
alcalinisation de l'électrolyte à proximité de la cathode. Cet effet est
favorisé par la présence du cation NH4+ qui diminue la
surtension de dégagement d'hydrogène. On peut supposer que la décharge de ce
cation est possible, dans le domaine de pH légèrement basique, suivant la
réaction:
NH4+ + e- è Hads + NH 3(aq)
L'hydrogène adsorbé
diffuse dans le métal et forme, par recombinaison, de l'hydrogène gazeux.
Globalement la
réaction devient:
![]()
2NH4+ + 2e- è 2
NH 3(aq) + H2
![]() Activation de la cathode
Activation de la cathode
En solution aqueuse neutre, le manganèse se
passive très rapidement. La couche d'oxyde qui se forme, ralentit puis bloque
la croissance du dépôt en augmentant la surtension cathodique. De ce fait,
l'élaboration du manganèse par électrolyse est impossible en l'absence de
dépassivation. Or, l'ammonium favorise l'autodissolution du manganèse et libère
la surface de l'électrode. Le manganèse peut alors se déposer plus facilement
avec une surtension cathodique plus faible. L'ajout de 2 moles l-1
de (NH4+)2SO4 à une solution 0,5M
de MnSO4 à pH 8,2 à 25°C, lors d'une électrolyses à 9,5 Adm-2,
modifie le potentiel cathodique de –1472 mV/ENH à –1193 mV/ENH. L'ammonium
introduit dans l'électrolyte, active ainsi la surface de l'électrode de
manganèse et rend possible la formation d'un dépôt.
![]() Conductivité du bain
Conductivité du bain
Enfin, la présence des sels d'ammonium
améliore la conductivité électrique de l'électrolyte qui doit permettre
l'utilisation de hautes densités de courant.
Dans la plupart des cas, l'électrolyse du
manganèse se déroule dans une cellule à compartiment double, l'anode et la
cathode étant séparées par un diaphragme. En effet, en absence de diaphragme,
l'acidification de l'électrolyte résultant des réactions qui ont lieu du côté
anodique, peut occasionner une diminution du pH à la cathode. Malgré l'effet
tampon du couple NH4+/NH3, le dégagement
cathodique d'hydrogène est ainsi favorisé par rapport à la réaction de
réduction des espèces manganèse.
En milieu sulfate, des cations manganeux
s'oxydent en Mn 4+ avec formation de dioxyde de manganèse qui peut
adhérer ou non à l'anode et être inclus dans le dépôt de manganèse. La réaction
d'oxydation s'écrit :
Mn 2+ + 2H2O è MnO2 + 4H+
+ 2 e –
En milieux chlorure, outre la formation de MnO2
, les anions chlorures s'oxydent en chlore:
2
Cl - è Cl2 + 2
e –
Une réaction secondaire est à l'origine d'un
dégagement d'azote :
![]()
2NH4+ + 3 Cl2 è N2 + 8
H+ + 6Cl-
Cette réaction est source d'acidification de
l'anolyte qui doit être séparé du catholyte par le diaphragme. De plus, cette
réaction consomme de l'ammonium, il faut donc ajuster sa concentration
régulièrement.
I ) 3
L'alliage zinc-manganèse
Les différentes phases constituées à partir du
zinc et du manganèse peuvent être présentées sur le diagramme de phase suivant
:
L'électrodéposition de l’alliage zinc-manganèse
a fait l’objet de plusieurs publications [16-23]. D'une manière générale les
bains utilisés étaient à base de sulfates de zinc et de manganèse dans du
citrate de sodium et étaient proposés avec des concentrations très
différentes :
ZnSO4 : 0,03 à 0,31 mol l-1 ;
MnSO4 : 0,09
à 0,36 mol l-1 ;
Na3C6H5O7 : 0,2 à 0,8 mol l-1
Les dépôts étaient préparés à température
ambiante ; le pH était compris entre 5 et 6. Des teneurs en manganèse de 5
à 100% étaient obtenues.
Une thèse a été initiée dans le laboratoire,
en collaboration avec Peugeot SA, pour tester la possibilité de préparer des
alliages zinc-manganèse à partir de ce type de bain. La composition choisie
était :
ZnSO4 : 0,24 mol l-1 ;
MnSO4 : 0,30 mol l-1 ;
Na3C6H5O7 : 0,61 mol l-1.
Plusieurs points ont été mis en évidence [1 et
ses références.]:
![]() Il est impossible de
préparer des alliages contenant plus de 12% de manganèse en conservant un
rendement compatible avec la réalité industrielle. L’augmentation du contenu en
Manganèse se fait au détriment du rendement, qui peut devenir inférieur à 20%.
Il est impossible de
préparer des alliages contenant plus de 12% de manganèse en conservant un
rendement compatible avec la réalité industrielle. L’augmentation du contenu en
Manganèse se fait au détriment du rendement, qui peut devenir inférieur à 20%.
![]() Pour obtenir des
alliages à 12% de Mn, le pH doit être contrôlé à moins d’une unité. La
condition 5,7 < pH <6,3 est incompatible avec des bains industriels.
Pour obtenir des
alliages à 12% de Mn, le pH doit être contrôlé à moins d’une unité. La
condition 5,7 < pH <6,3 est incompatible avec des bains industriels.
Cependant, l’ensemble des résultats publiés
sur le comportement à la corrosion de l’alliage zinc-manganèse indique une
résistance à la corrosion nettement supérieure à celle du zinc pur. Cette bonne
résistance à la corrosion, testée par la méthode du brouillard salin, est
obtenue pour des alliages contenant au moins 50% de manganèse, la valeur
optimum en manganèse étant de 66%, avec une amélioration de la résistance dés
40%. Il faut souligner qu’aucun résultat de corrosion naturelle n’est indiqué.
Cette réputation de bonne tenue à la corrosion nous a incité à chercher de
nouveaux bains permettant de préparer des dépôts zinc-manganèse, avec des
paramètres moins contraignants.
I ) 4 Généralités sur l'électrocristallisation
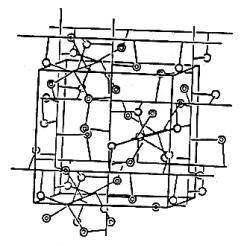
I ) 4.1 Aspects généraux de l’électrocristallisation des métaux
L'électrocristallisation est due à un
transfert de charge entre un cation métallique, le plus souvent solvaté, et un
substrat conducteur. Les autres ions ou molécules présents dans la solution,
susceptibles de s'adsorber sur le substrat, font de l'électrocristallisation un
processus complexe qui fait intervenir un grand nombre de paramètres. Le
processus d'électrocristallisation est fonction d'un certain nombre de
variables dont les principales sont présentées sur le schéma suivant [24]:
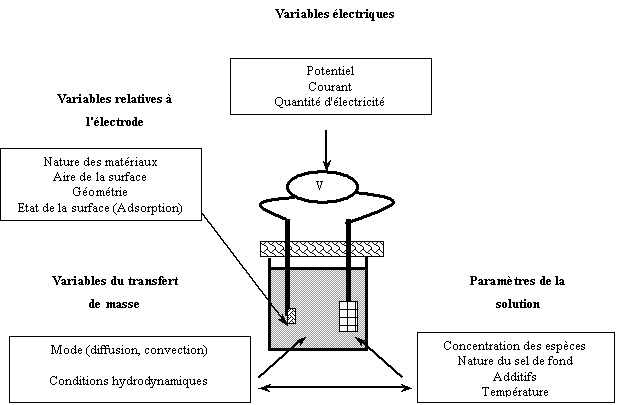
Principales variables du processus d'électrocristallisation.
I ) 4.2 Aspects thermodynamiques et cinétiques [25]
Le processus d’électrocristallisation peut se
décomposer en deux processus principaux : la germination et la croissance des
germes formés.
L'électrocristallisation du métal se produit à
l’interface d’un substrat conducteur électronique et d’une solution (conducteur
ionique). En général, elle comprend trois étapes:
i)
La formation d’adatomes métalliques, Me adsorbé , sur le même substrat
métallique Me, ou sur un autre type de substrat S.
ii)
La formation d’une phase deux dimensions (2D) et/ou trois dimensions (3D) après
une phase de germination et de croissance de germes.
iii)
La croissance en 3D du dépôt de Me.
Le substrat S, différent du métal Me, est
considéré comme électrochimiquement inactif dans la zone de potentiel étudiée.
Deux facteurs principaux déterminent le
processus électrochimique de cristallisation d'un métal.
![]() Le premier concerne les
aspects thermodynamiques de la croissance des phases 2D et 3D qui peuvent être
traités de la même manière que la déposition de Me à partir d’une phase vapeur.
Le premier concerne les
aspects thermodynamiques de la croissance des phases 2D et 3D qui peuvent être
traités de la même manière que la déposition de Me à partir d’une phase vapeur.
![]() Le second concerne
l’électrolyte dont dépendent fortement la structure de l’interface
substrat/électrolyte, la cinétique de transfert de charge et de masse, la
cinétique des réactions chimiques qui précèdent ou qui succèdent au transfert
de charge.
Le second concerne
l’électrolyte dont dépendent fortement la structure de l’interface
substrat/électrolyte, la cinétique de transfert de charge et de masse, la
cinétique des réactions chimiques qui précèdent ou qui succèdent au transfert
de charge.
Le domaine de stabilité des phases 2D et 3D
sur S est caractérisé par l’équation de Nernst, qui décrit l’équilibre
thermodynamique d’une cristallisation ou d’une dissolution d’un métal Me, sur
lui même ou sur un substrat différent S. Pour la formation et la croissance 3D
d’un dépôt de Me sur lui même ou sur un autre substrat, la réaction globale du
couple Mez+/Me est :
Mez+solv
+ z e- ó Me (1)
où Mez+ solv représente
l’ion métallique solvaté dans l’électrolyte.
L’équilibre de Nernst du couple Mez+ /Me
est donné par:
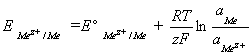 (2)
(2)
E Mez+ /Me est le
potentiel d’équilibre de Nernst du couple Mez+/Me. E° Mez+/Me
correspond au potentiel standard du couple Mez+/Me et a Mez+
désigne l’activité des ions Mez+solv dans l’électrolyte. Pour un dépôt de Me, a Me
= 1.
Le potentiel d’électrode, E, détermine le sens
des réactions de (1). Un dépôt Me peut être déposé cathodiquement pour E<E Mez+/Me.
Par contre, le dépôt ne se forme pas pour E>EMez+/Me.
Par conséquent le potentiel d’équilibre de Nernst représente la limite
supérieure du domaine de stabilité du dépôt.
La surtension de cristallisation, hc , du couple Mez+/ Me, est définie par:
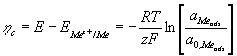 (3)
(3)
où aMe ads et a0, Me ads
représentent respectivement les activités de Meads pour E¹EMez+/Me et E=EMez+/Me.
Cette définition implique que toutes les étapes qui se produisent durant
l’étape (1) comme le transfert de masse, la diffusion, et les réactions
chimiques sont suffisamment rapides afin qu’elles ne rentrent pas en compte
dans l’équilibre chimique. Alors leurs contributions à la surtension deviennent
négligeables, on a htotal = hc . La cristallisation de Me se produit avec une surtension cathodique, hc < 0, c’est à dire en sursaturation Dm>0. La dissolution de Me
se produit par contre pour des surtensions anodiques, hc > 0, c’est à dire en sous saturation Dm<0. La sursaturation ou
sous saturation Dm sont définies par :
Dm= m Me ads - m Me 0 ,ads = -z F ( E - E Mez+ /Me) (4)
En utilisant la définition du potentiel
chimique mi = mi° + (RT) ln ai
où mi° représente le potentiel chimique standard du composé i.
Dans le cas de la formation d'un dépôt sur un
substrat différent S, la globalité de la réaction du couple S/Mez+
devient :
Mez+sol
+ z e- ó Me(à la surface de S) (5)
Pour un dépôt de Me en surface de S,
l'équilibre thermodynamique est donné par l'équation de Nernst (2). Le
potentiel d'électrode, E, détermine le sens de la réaction. Cette affirmation
est vérifiée tant que le potentiel du dépôt de Me est inférieur au potentiel de
stabilité, E<EMez+/Me. Dans certaines conditions, une
phase 2D est stable pour E>EMez+/Me et Dm<0 (sous
saturation) sur un substrat différent. Par conséquent, la différence de
potentiel E- EMez+/Me peut être définie, par convention,
comme :
![]() DE (sous potentiel)
> 0 pour E > EMez+/Me
DE (sous potentiel)
> 0 pour E > EMez+/Me
E- EMez+/Me
= (6)
h (sur
potentiel) < 0 pour E < EMez+/Me
Dans ce cas, le potentiel d'équilibre de
Nernst, EMez+/Me, représente la limite du domaine de
stabilité à la fois de la phase 2D et du dépôt de Me. Pour E= EMez+/Me
, la phase 2D et le dépôt de Me coexistent. Par conséquent, une cristallisation
en sous potentiel (UPD) et en sur potentiel (OPD) de Me sur S sont liées à la
formation d'une phase 2D puis 3D. Dans le cas d'un UPD de Me à E> EMez+/Me
, hc doit être remplacé par DE dans l'équation (3)
en accord avec l'équation (6).
Le processus d'OPD de Me sur S devient
identique avec l'OPD de Me sur Me, si le film de Me déposé dépasse une certaine
épaisseur et se comporte comme une phase 3D. Alors l'équation (5) devient
identique à l'équation (1). Généralement, le domaine d'épaisseur critique est
compris entre 1 et 20 monocouches environ de Me sur S.
I ) 4.3 Mécanismes de cristallisation des métaux [25]
Les paramètres déterminants du mécanisme d'UPD
et d'OPD du métal sur un substrat différent sont : l'énergie de liaison Meads-S
et les différences des paramètres cristallographiques entre S et le dépôt 3D.
En considérant le processus d'électrocristallisation, à des conditions proches
de l'équilibre, (faible sursaturation et effets cinétiques négligeables), et en
ne tenant pas compte de la formation d'alliages Me-S, deux modes différents de
croissance peuvent être schématiquement illustrés.
i)
Dans le premier cas (a), l'énergie de
liaison de Meads sur le substrat S , YMe ads-S, est inférieure à celle de Me sur le substrat Me, Y Me
ads-Me. Par conséquent, la concentration en surface
de Meads à EMez+/Me est faible et l'OPD de la
phase 3D se produit sur une surface de substrat non modifiée, en suivant le
mode de croissance "Volmer-Weber" (ou un mode de croissance d'îlot)
moins sensible aux différences de paramètres cristallographiques entre Me et S.
ii)
Dans le second cas, l'énergie de liaison
de Meads sur le substrat S ,Y Me ads-S, est supérieure à celle de
Me sur le substrat Me, Y Me ads-Me. Alors, la phase 2D peut être
formée dans le domaine de l'UPD et la concentration à la surface de Meads
à EMez+/Me peut atteindre une ou plusieurs monocouches de
Meads en fonction de Y Me ads-S.
Deux sous catégories peuvent être alors distinguées.
ii1)
Pour les systèmes où la différence des paramètres cristallographiques Me-S est
faible, les couches 2D formées en UPD et les cristallites 3D de Me ont une
croissance épitaxique orientée, qui suit le mode de croissance "Frank-van
der Merwe" (ou mode de croissance couche par couche) jusqu'à la disparition
de la forte attraction Me-S. Par la suite le nombre de couches de Me étant
suffisant, la croissance continue de la même façon que sur le substrat Me.
ii2)
Pour les systèmes où la différence des paramètres cristallographiques Me-S est
significative, la phase 2D formée dans le domaine de l'UPD présente une
structure différente de la structure 3D de Me, elle est soumise ainsi à de
fortes tensions internes. La formation et la croissance de cristallites 3D sans
tension interne (îlots), sur une couche 2D contrainte, suivent le mode de
croissance "Stranski-Krastanov" le plus énergétiquement favorable. La
différence des paramètres cristallographiques entre la phase 2D de Meads
et du dépôt 3D est principalement résolue par la formation de dislocations.


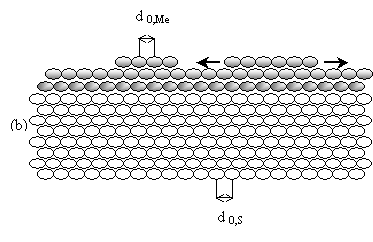
(b)
Mode de croissance "Frank-van der Merwe" (formation couche par couche) pour
Y Me
ads-S>> Y Me ads-Me, et un rapport (d0,Me - d0,S)/
d0,S » 0.
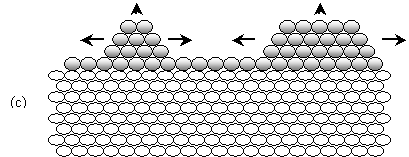
(c)
Mode de croissance "Stranski-Krastanov" (formation
d'îlots 3D précédé par une couche 2D Meads sur S) pour
Y Me
ads-S>> Y Me ads-Me, et un rapport (d0,Me - d0,S)/
d0,S > 0 ou (d0,Me - d0,S)/ d0,S
< 0.
I ) 4.4 Electrocristallisation d'alliages
Théoriquement, l'électrocristallisation
d'alliages résulte d'une codéposition d'au moins deux métaux pouvant
cristalliser dans une des phases du diagramme de phases. Cependant, certaines
conditions d'électrocristallisation favorisent les solutions de métaux réputés
insolubles. L'électrocristallisation rend possible l'existence de phases hors
de leur domaine de stabilité thermique ainsi que celle de composés intermétalliques
inconnus par ailleurs. Dans certains cas les alliages électrodéposés peuvent
être obtenus avec une finesse de grains pouvant aller jusqu'à une structure
amorphe.
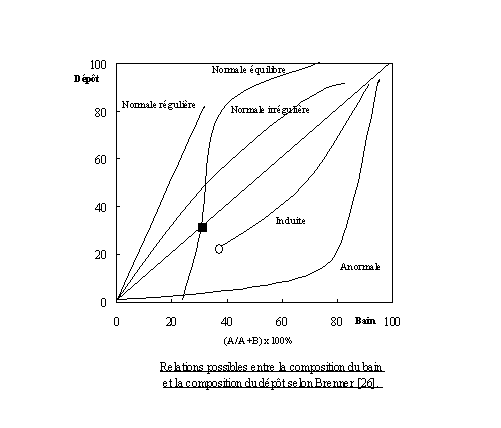
Cependant, l'électrocristallisation des
alliages est plus complexe à étudier que celle des métaux purs. En effet, on
peut supposer qu'un adatome d'une espèce peut agir comme un inhibiteur ou comme
catalyseur pour la réduction du métal de l'autre espèce. Dans le premier cas où
la réduction du métal le plus noble est inhibé, on qualifie la codéposition
d'anormale, selon la classification de Brenner [26]. Dans le second cas, la
cristallisation d'un métal rend possible la réduction d'un autre métal, qui ne
se réduit pas seul en solution, on qualifie alors la codéposition d'induite.
Dans la plupart des cas les potentiels d'électrocristallisation sont trop
éloignés pour permettre la formation d'un alliage. L'utilisation de substances
organiques (les additifs) permet de rapprocher les potentiels et d'obtenir des
codépôts sous forme d'alliages.
I ) 5 Utilisation des additifs
Les procédés électrolytiques modernes
utilisent, en fonction des besoins, des bains dont les compositions sont très
spécifiques. Cette spécificité est fonction du ou des métaux à électrodéposer
et des caractéristiques que l'on veut conférer au dépôt. La formulation de ces
bains constitue un intérêt constant tant de la part des industriels que des
chercheurs. Pour les premiers, le rendement d'un procédé est directement lié à
la composition du bain: l'élaboration d'un bain performant constitue en soit un
enjeu économique et les compositions de très nombreux bains font l'objet de
brevets. La relation qui existe entre la nature du dépôt et la composition du
bain est très difficile à établir, il n’existe pas de règles générales, tout le
"savoir" est basé sur des observations empiriques. Ce domaine
constitue ainsi un champ d'étude important comme le démontre le nombre de
publications sur ce sujet.
Les bains électrolytiques contiennent
généralement, outre les espèces métalliques à réduire, des additifs constitués
de mélanges de substances organiques ou parfois de sels minéraux. Malgré leur
faible concentration dans les bains, le rôle des additifs est déterminant. En
général, les additifs interviennent en favorisant ou en bloquant la réduction
des espèces électroactives. Ils permettent de maîtriser le procédé
d'électrocristallisation (régulation de l'électrocristallisation) et donc la
qualité du dépôt (contrôle des propriétés physiques comme la structure, la
dureté, la ductilité, la brillance etc.).
I ) 5.1 Principe de l’action d'un additif [27-32]
Malgré la grande diversité des additifs, une
revue bibliographique permet de distinguer trois types de mécanismes. L’additif
peut agir soit par :
1) Blocage de surface
Principe :
L’additif s’adsorbe sur la surface et bloque totalement certains sites de
transfert de charge.
Sans Additif Avec Additif
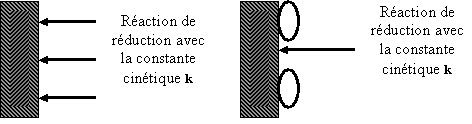 S
S
U
B
S
T
R
Molécule d’additif adsorbé
A
T
En présence d'additif, la constante cinétique
globale n'est pas modifiée. Cependant la réduction se fait sur une aire
réduite. Le mécanisme de cristallisation n’est pas modifié mais la quantité des
espèces électroactives réduites est directement fonction du taux de
recouvrement q de l'additif. Ce
paramètre est lui même fonction de la nature du substrat, de l’affinité de
chaque molécule avec la surface (pour des raisons soit géométriques, soit
électroniques ...), du pH, du potentiel imposé, de la température etc. .
2) Adsorption réactive
Principe :
L’additif s’adsorbe à la surface et modifie la cinétique de réaction sur
certains sites.

Outre la réduction avec la constante cinétique
k, la molécule d'additif peut ralentir ou accélérer la réaction de réduction.
Une seconde constante est introduite qui confère à la réaction une constante
cinétique globale:
kt= k’(q) + k
(1-q)
L’additif peut agir sur la constante cinétique
selon les cas par modification du potentiel d’Helmholtz, par effet de pontage
(catalyse), par modification de tension interfaciale.
3) Complexation en solution
Principe:
L’additif complexe l’espèce électroactive en solution.
Dans ce cas c’est l’étape de décomplexation
qui introduit une nouvelle constante cinétique avant la formation de l’adatome.
I ) 5.2 Classification des additifs [27 et ses références]
Les additifs utilisés industriellement
contiennent généralement un mélange de sels minéraux et de molécules
organiques. Parmi ces additifs, on distingue différents types en fonction de
leur nature et de leurs effets.
![]() Les cations inorganiques
non réductibles à la cathode (Na+, K+, Mg2+,
Ca2+, Al3+) qui ont des effets très faibles.
Les cations inorganiques
non réductibles à la cathode (Na+, K+, Mg2+,
Ca2+, Al3+) qui ont des effets très faibles.
![]() Certains anions
inorganiques qui peuvent modifier la double couche et par conséquent la
surtension de transfert de charge en l'activant (Cl-, Br-,
I-) ou en l'inhibant (BF4-, NH2SO22-,
ClO4-).
Certains anions
inorganiques qui peuvent modifier la double couche et par conséquent la
surtension de transfert de charge en l'activant (Cl-, Br-,
I-) ou en l'inhibant (BF4-, NH2SO22-,
ClO4-).
![]() Les espèces organiques
dont les effets en solution sont variables. Elles sont fortement inhibantes si
elles sont adsorbées sur le métal et si elles ne montrent pas d'affinité avec
l'eau. Cette adsorption est fonction de la nature et de la dimension des
molécules, mais le critère déterminant est sans doute la structure
électronique. En effet, les composés qui présentent une adsorption maximale sur
le zinc, ont une énergie d'ionisation d'environ 9,4 eV qui correspond à
l'énergie d'ionisation du couple Zn/Zn2+.
Les espèces organiques
dont les effets en solution sont variables. Elles sont fortement inhibantes si
elles sont adsorbées sur le métal et si elles ne montrent pas d'affinité avec
l'eau. Cette adsorption est fonction de la nature et de la dimension des
molécules, mais le critère déterminant est sans doute la structure
électronique. En effet, les composés qui présentent une adsorption maximale sur
le zinc, ont une énergie d'ionisation d'environ 9,4 eV qui correspond à
l'énergie d'ionisation du couple Zn/Zn2+.
Les additifs peuvent être également classés en
fonction de leurs effets sur les dépôts. Dans ce cas, nous pouvons les classer
en deux grandes familles: les additifs ayant un effet nivelant et les additifs
ayant un effet brillanteur.
I ) 5.3 Effet nivelant des additifs [33]
 L'élimination des irrégularités de surface est un des effets
macroscopiques intéressant apporté par les additifs. On appelle cet effet
:"effet nivelant". Ce processus est une résultante directe de
l'action catalytique des additifs. En effet, la concentration des espèces
activantes est différente sur une surface concave ou convexe en fonction des
effets de champ. Ainsi la vitesse de cristallisation augmente dans les trous
(surface concave) et diminue sur les bosses (surface convexe). Il en résulte
une mise à niveau immédiate de la surface. Ce mécanisme est illustré par la
figure suivante.
L'élimination des irrégularités de surface est un des effets
macroscopiques intéressant apporté par les additifs. On appelle cet effet
:"effet nivelant". Ce processus est une résultante directe de
l'action catalytique des additifs. En effet, la concentration des espèces
activantes est différente sur une surface concave ou convexe en fonction des
effets de champ. Ainsi la vitesse de cristallisation augmente dans les trous
(surface concave) et diminue sur les bosses (surface convexe). Il en résulte
une mise à niveau immédiate de la surface. Ce mécanisme est illustré par la
figure suivante.
I ) 5.4 Effet brillanteur des additifs
L'usage d'additifs permet également d'obtenir
des dépôts brillants. Cette caractéristique est obtenue si le dépôt remplit
deux critères. La taille des cristallites qui le compose doit être inférieure à
la longueur d'onde de la lumière visible (0,4 mm) et d'autre part, le dépôt
doit présenter une structure de grains orientée. Mais le premier critère n'est
pas suffisant car la brillance dépend du degré d'orientation dans le même plan
[27].
Les additifs entraînent l'augmentation de la
finesse des grains en multipliant le nombre de germes de cristallisation. En
s'adsorbant sur le substrat, ils créent de nouveaux centres d'incorporation en
limitant la diffusion en surface des adatomes.
I ) 5.5 Effet synergique des additifs
En général, l'emploi de molécules isolées
conduit à un dépôt de qualité insuffisante. Par contre en présence d'une autre
molécule l'effet sur le dépôt peut devenir remarquable par effet de synergie entre les additifs
[27]. L'exemple suivant illustre un effet synergique des additifs, identifié
sous le terme de "modèle de perforation locale".
L'utilisation d'un surfactant seul dans le
bain électrolytique bloque le processus de réduction des espèces électroactives
en formant un film sur la surface cathodique. La formation d'un dépôt devient
alors impossible sans augmenter la surtension cathodique. Or, le simple ajout
d'une petite quantité de "mouillant" permet d'activer le processus de
réduction. Le dépôt obtenu possède alors une structure microcristalline avec un
grain fin et une surface brillante [33]. C'est la présence du mouillant qui,
fortement lié à la surface du substrat, empêche le surfactant de recouvrir la
totalité de la surface. L'apparition de ces distorsions permet aux ions de
pénétrer sous le film et de se réduire. Le mécanisme de croissance du dépôt
n'est alors plus contrôlé par les paramètres classiques de
l'électrocristallisation mais par l'action combinée des deux additifs.
Le modèle de perforation locale peut être
illustré par le schéma suivant:
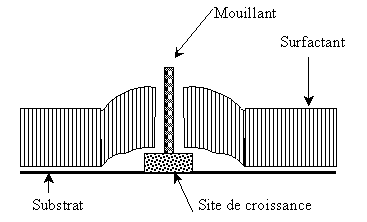
Modèle
de perforation locale montrant l'effet synergique de l'action du brillanteur
et du surfactant [33].
I ) 5.6 Influence des additifs sur la morphologie des dépôts [34]
La morphologie du cristal résulte d'une
compétition entre la croissance latérale et la croissance normale au substrat.
La croissance normale est fortement liée à la fréquence de nucléation à deux
dimensions. Les observations montrent que l'épaisseur des marches de croissance
augmente avec la densité du courant, mais décroît quand l'intensité
d'inhibition augmente. Ainsi, quand la densité de courant augmente, la vitesse
de croissance latérale diminue, ce qui favorise la formation de cristallites
isolées ou de dendrites. A l'opposé, quand l'inhibition augmente, la vitesse de
croissance latérale augmente favorisant un dépôt dense et cohérent. Le mode de
croissance des dépôts est donc directement corrélé à ces deux facteurs. Cette
approche pragmatique permet d'établir une relation entre les caractéristiques
morphologiques des dépôts et certains paramètres physiques mesurables. Dans le
cas de l'électrocristallisation, la surtension cathodique constitue un
paramètre déterminant. Cependant les difficultés expérimentales ne permettent
pas d'accéder à ce paramètre, ni à ces différentes composantes : surtension de
transfert de charge, surtension de diffusion, surtension de réaction,
surtension de cristallisation. Nous devons donc considérer deux autres
paramètres importants: le rapport de la densité de courant cathodique sur la
concentration des espèces métalliques J/ CMe z+ et
l'intensité d'inhibition. Contrairement au premier paramètre, il convient de
définir la notion d'intensité d'inhibition. D'après Fischer [34], l'inhibition
est due à la présence de substances (molécules, atomes, ou ions) à la surface
de l'électrode, dans la couche d'Helmholtz ou dans la couche de diffusion. Ces
additifs entravent les processus cathodiques, modifient la surtension, la
structure métallographique et la texture des dépôts. Ils sont chimiquement ou
physiquement adsorbés sur une partie de la surface cathodique ou des sites
électroactifs. La sensibilité du métal aux additifs est d'autant plus grande
que l'affinité du métal pour l'additif augmente, et que le coefficient de
diffusion décroît. Bien que l'intensité de l'inhibition soit un facteur
difficile à quantifier, nous pouvons toutefois le relier à la concentration de
l'inhibiteur en solution ou en surface. Ainsi différents modes de croissance
des dépôts sont établis en fonction de la combinaison des deux paramètres J/ CMe z+ et
intensité d'inhibition.
Fischer propose cinq modes principaux de
croissance de dépôt polycristallin à partir d'observations métallographiques:
(1) Type (DI) formant un domaine de cristallites isolées orientées
(2) Type (RB) reproduisant le modèle de base orienté
(3) Type (Z) intermédiaire
(4) Type (DT) formant un domaine texturé orienté
(5) Type (D) avec une dispersion sans orientation.
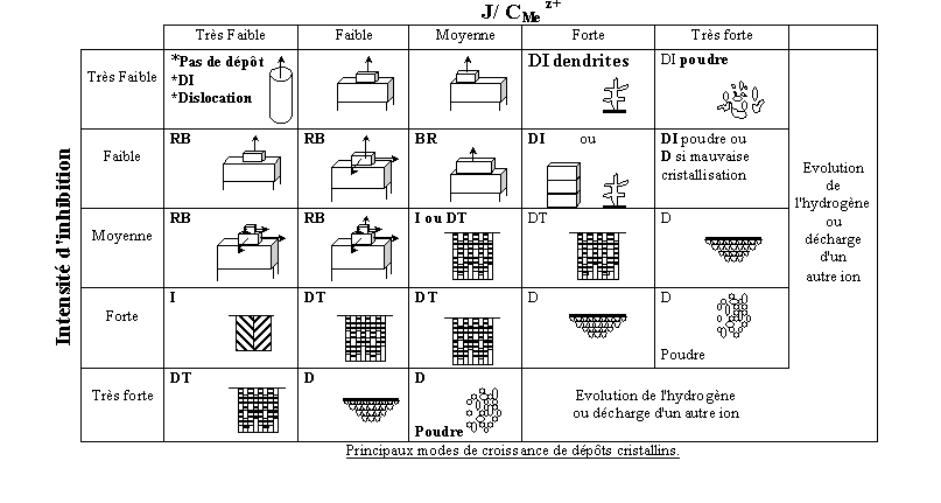
Le premier type (DI) est habituellement
observé pour une faible inhibition. En augmentant la densité de courant, on
obtient successivement des cristaux prismatiques, des dendrites puis finalement
de la poudre. Le second type (RB) est observé pour une inhibition et une
densité de courant modérée. La vitesse de croissance latérale permet une bonne
croissance des cristallites. Si la durée d'électrolyse est importante, la rugosité
du dépôt augmente et le type (RB) peut se dégrader en type (DI). Le troisième
type est considéré par Fischer comme l'intermédiaire entre le deuxième (RB) et
le quatrième type (DT). Le quatrième type (DT) est observé pour une forte
inhibition et/ou densité de courant, il est caractérisé par un grand nombre de
cristallites allongées perpendiculairement au substrat qui forment un dépôt
cohérent. Le cinquième type (D) est obtenu pour une très forte inhibition et/ou
densité de courant. Il est formé par un grand nombre de petites cristallites
qui constituent un dépôt cohérent. Les quatre premiers types de dépôts sont
obtenus à partir d'une nucléation à deux dimensions alors que le cinquième type
nécessite une nucléation à trois dimensions. Les principaux types de nucléation
sont présentés dans le tableau précédent qui montre les domaines de stabilité.
La figure suivante en est une version simplifiée.